- MACROMOLÉCULES
- MACROMOLÉCULESLes macromolécules sont des systèmes moléculaires constitués par un très grand nombre d’atomes assemblés entre eux par des liaisons covalentes. Alors que les molécules simples sont formées de quelques dizaines d’atomes au maximum, les macromolécules se caractérisent par des dimensions moléculaires et des masses molaires très largement supérieures.Le terme «macromolécule» est général et se rapporte, en principe, à tout système défini comme ci-dessus, qu’il soit organique, inorganique, naturel, artificiel ou synthétique; cependant, il est préférentiellement employé pour décrire des systèmes organiques et même, dans cette catégorie, plutôt ceux d’origine naturelle.Les macromolécules artificielles, qui résultent de la modification chimique de macromolécules naturelles, ainsi que les macromolécules synthétiques, créées de toutes pièces à partir de molécules simples, sont le plus souvent appelées polymères. «Macromolécule» est cependant un terme bien adapté à la très grande variété des assemblages moléculaires qui, à présent, peuvent être obtenus, et c’est donc sous cet intitulé que seront développés les aspects physico-chimiques et chimiques de ces composés, les aspects physiques étant développés dans l’article POLYMÈRES.La science macromoléculaire est relativement récente, puisque c’est seulement dans les années 1920 que H. Staudinger (qui reçut le prix Nobel de chimie en 1953) a proposé la notion de macromolécule, qui s’opposait alors à la théorie micellaire (théorie selon laquelle les grosses molécules étaient interprétées comme une agrégation de petites molécules reliées entre elles par des liaisons non covalentes). La théorie macromoléculaire a triomphé et l’importance économique des matériaux polymères a suscité, à partir de la fin des années 1930, une explosion des recherches dans ce domaine, aussi bien théoriques qu’industrielles. En effet, les composés macromoléculaires sont présents dans tous les secteurs de l’activité économique, essentiellement sous forme de matériaux de structure, mais aussi dans les domaines du génie biomédical, de l’opto-électronique, du traitement des eaux résiduaires...La plupart des propriétés particulières des composés macromoléculaires sont le reflet de la multiplicité des interactions moléculaires qui se développent soit à l’intérieur d’une même chaîne, soit entre les différentes chaînes constitutives d’un système donné. Il en résulte que ces propriétés sont fortement dépendantes de la nature des groupements moléculaires qui constituent une chaîne macromoléculaire.1. Structure des macromoléculesLe terme «structure» recouvre ici différents niveaux, qu’il est important d’expliciter.La dimensionnalité d’une macromolécule est une propriété structurale qui influence fortement ses propriétés physiques. Les macromolécules monodimensionnelles, les plus simples, présentent une architecture vermiculaire dont les anneaux successifs correspondent aux motifs constitutifs (ou unités monomères). C’est la répétition de ces motifs qui engendre la macromolécule. La famille des macromolécules monodimensionnelles inclut toutes celles dont les dimensions moléculaires sont finies (masses molaires de quelques milliers à quelques millions de g.mol—1), qu’elles soient linéaires ou ramifiées.Les macromolécules bidimensionnelles développent des liaisons covalentes qui unissent les motifs constitutifs dans deux dimensions de l’espace et forment ainsi des feuillets monomacromoléculaires. De telles architectures sont difficiles à synthétiser; elles sont rares et structuralement imparfaites dans la famille des polymères organiques naturels. On les rencontre plus fréquemment parmi les polymères minéraux (talc, mica...).Les macromolécules tridimensionnelles, comme les bidimensionnelles, résultent d’un lien de valence supérieur à 2 entre certains motifs constitutifs d’un ensemble macromoléculaire. Il en résulte la formation de systèmes monomacromoléculaires dont les dimensions sont celles de l’objet qu’ils constituent; les masses molaires correspondantes peuvent être considérées comme infinies. La description moléculaire des réseaux macromoléculaires tridimensionnels fait appel à la longueur moyenne (caractérisée par la masse molaire moyenne) des arêtes comprises entre deux nœuds ou à celle des mailles.L’enchaînement des motifs constitutifs d’une macromolécule synthétique est obtenu par une réaction dite de polymérisation. Celle-ci peut faire intervenir un ou plusieurs types de molécules monomères, dont la structure va déterminer celle des motifs constitutifs (unités monomères) de la chaîne. Ainsi, les molécules monomères génèrent les unités monomères. Les macromolécules issues d’un seul type de molécules monomères sont appelées homopolymères, quelle que soit la structure des unités monomères qui en résultent. Lorsque les unités répétitives d’un polymère sont identiques et s’enchaînent de façon régulière, l’homopolymère est dit régulier; il est irrégulier si la régiosélectivité de l’enchaînement (localisation des liens de valence sur chaque unité monomère) n’est pas respectée.Lorsqu’un système macromoléculaire est formé à partir de plusieurs types de molécules monomères, il est qualifié de copolymère. Si A et B représentent deux unités monomères différentes, elles peuvent être enchaînées de différentes façons pour former des macromolécules.Les copolymères statistiques, sont des copolymères dans lesquels les différentes unités monomères sont réparties statistiquement, par exemple:
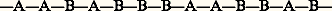 Ils présentent généralement des caractéristiques intermédiaires entre celles des homopolymères polyA et polyB qui auraient été obtenus par polymérisation séparée des unités monomères A et B. Ces caractéristiques sont déterminées par la composition en A et en B du copolymère et par la fréquence des alternances —AB— ou —BA— le long de la chaîne. Les copolymères à blocs polyA et polyB:
Ils présentent généralement des caractéristiques intermédiaires entre celles des homopolymères polyA et polyB qui auraient été obtenus par polymérisation séparée des unités monomères A et B. Ces caractéristiques sont déterminées par la composition en A et en B du copolymère et par la fréquence des alternances —AB— ou —BA— le long de la chaîne. Les copolymères à blocs polyA et polyB: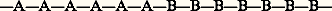 présentent des propriétés nouvelles, sensiblement différentes de celles des homopolymères correspondants; il en est de même pour les copolymères greffés, qui comportent une chaîne principale, formée par l’une des branches monomères avec des unités constituées par des blocs de l’autre monomère.Dans le cas des homopolymères réguliers, qui présentent un élément d’asymétrie (le plus souvent un carbone tertiaire ou quaternaire) pour chaque unité monomère, il est intéressant de considérer les configurations relatives des éléments d’asymétrie successifs. Lorsqu’une chaîne est majoritairement formée d’unités successives de même configuration, le polymère correspondant est qualifié d’isotactique. Lorsque les configurations relatives successives sont majoritairement opposées, il est dit syndiotactique. En cas d’absence de régularité configurationnelle, le polymère est atactique. L’ordonnancement de la succession des motifs configurationnels des polymères, ou tacticité, est mesuré par la proportion de triades (ensemble de trois motifs successifs) iso-, syndio- ou hétérotactiques du système macromoléculaire considéré.Le polypropène, ou polypropylène, isotactique est l’un des polymères synthétiques les plus intéressants au plan économique, pour ses propriétés mécaniques et pour son faible coût de production.Un autre type d’isomérie configurationnelle doit être considéré pour les homopolymères présentant des doubles liaisons dans la chaîne. Ainsi, le caoutchouc naturel est l’isomère cis du 1,4-polyisoprène,
présentent des propriétés nouvelles, sensiblement différentes de celles des homopolymères correspondants; il en est de même pour les copolymères greffés, qui comportent une chaîne principale, formée par l’une des branches monomères avec des unités constituées par des blocs de l’autre monomère.Dans le cas des homopolymères réguliers, qui présentent un élément d’asymétrie (le plus souvent un carbone tertiaire ou quaternaire) pour chaque unité monomère, il est intéressant de considérer les configurations relatives des éléments d’asymétrie successifs. Lorsqu’une chaîne est majoritairement formée d’unités successives de même configuration, le polymère correspondant est qualifié d’isotactique. Lorsque les configurations relatives successives sont majoritairement opposées, il est dit syndiotactique. En cas d’absence de régularité configurationnelle, le polymère est atactique. L’ordonnancement de la succession des motifs configurationnels des polymères, ou tacticité, est mesuré par la proportion de triades (ensemble de trois motifs successifs) iso-, syndio- ou hétérotactiques du système macromoléculaire considéré.Le polypropène, ou polypropylène, isotactique est l’un des polymères synthétiques les plus intéressants au plan économique, pour ses propriétés mécaniques et pour son faible coût de production.Un autre type d’isomérie configurationnelle doit être considéré pour les homopolymères présentant des doubles liaisons dans la chaîne. Ainsi, le caoutchouc naturel est l’isomère cis du 1,4-polyisoprène,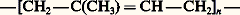 ,alors que la gutta-percha est l’isomère trans. Ces deux polymères naturels, d’isomérie configurationnelle opposée, montrent des propriétés nettement différenciées.Comme toutes les molécules, les macromolécules ont tendance à adopter une géométrie correspondant à l’état énergétique le plus stable; en particulier, si leur environnement le permet, elles vont adopter, par rotation autour des liaisons simples du squelette de la chaîne, des conformations qui favorisent au maximum les interactions et minimisent les répulsions. Lorsque les irrégularités des enchaînements ou des configurations empêchent le développement régulier des interactions ou bien lorsque celles-ci sont rompues sous l’effet d’un solvant ou de la température, les liens intra- ou intermoléculaires s’établissent de façon désordonnée. Il en résulte la formation de pelotes statistiques (de structure vermiculaire).En revanche, lorsque toutes les conditions de régularité structurale en amont sont réunies, les macromolécules ont tendance à donner des conformations régulières correspondant soit à un zigzag planaire, soit à une structure hélicoïdale de la chaîne principale (structure des protéines, par exemple).Cependant, même pour des structures moléculaires très régulières, l’énergie nécessaire à la perfection de la conformation est trop élevée pour être atteinte; les chaînes adoptent alors un état structural intermédiaire dans lequel alternent des séquences de conformation régulière et des séquences irrégulières sous la forme de pelotes statistiques.Toutes les propriétés des systèmes macromoléculaires sont influencées par les dimensions moléculaires. Hormis les enzymes et une famille de polymères récemment synthétisés par ingénierie génétique, tous les composés macromoléculaires sont polymoléculaires, c’est-à-dire constitués d’entités de dimensions moléculaires différentes. Cette polymolécularité implique l’utilisation de valeurs moyennes pour la description dimensionnelle de ces systèmes.Si Ni est le nombre de moles d’espèces de masse molaire Mi , les masses molaires moyennes en nombre (face="EU Upmacr" 聾n), en masse (face="EU Upmacr" 聾w 聾w) et viscosimétrique (face="EU Upmacr" 聾v), s’écrivent:
,alors que la gutta-percha est l’isomère trans. Ces deux polymères naturels, d’isomérie configurationnelle opposée, montrent des propriétés nettement différenciées.Comme toutes les molécules, les macromolécules ont tendance à adopter une géométrie correspondant à l’état énergétique le plus stable; en particulier, si leur environnement le permet, elles vont adopter, par rotation autour des liaisons simples du squelette de la chaîne, des conformations qui favorisent au maximum les interactions et minimisent les répulsions. Lorsque les irrégularités des enchaînements ou des configurations empêchent le développement régulier des interactions ou bien lorsque celles-ci sont rompues sous l’effet d’un solvant ou de la température, les liens intra- ou intermoléculaires s’établissent de façon désordonnée. Il en résulte la formation de pelotes statistiques (de structure vermiculaire).En revanche, lorsque toutes les conditions de régularité structurale en amont sont réunies, les macromolécules ont tendance à donner des conformations régulières correspondant soit à un zigzag planaire, soit à une structure hélicoïdale de la chaîne principale (structure des protéines, par exemple).Cependant, même pour des structures moléculaires très régulières, l’énergie nécessaire à la perfection de la conformation est trop élevée pour être atteinte; les chaînes adoptent alors un état structural intermédiaire dans lequel alternent des séquences de conformation régulière et des séquences irrégulières sous la forme de pelotes statistiques.Toutes les propriétés des systèmes macromoléculaires sont influencées par les dimensions moléculaires. Hormis les enzymes et une famille de polymères récemment synthétisés par ingénierie génétique, tous les composés macromoléculaires sont polymoléculaires, c’est-à-dire constitués d’entités de dimensions moléculaires différentes. Cette polymolécularité implique l’utilisation de valeurs moyennes pour la description dimensionnelle de ces systèmes.Si Ni est le nombre de moles d’espèces de masse molaire Mi , les masses molaires moyennes en nombre (face="EU Upmacr" 聾n), en masse (face="EU Upmacr" 聾w 聾w) et viscosimétrique (face="EU Upmacr" 聾v), s’écrivent: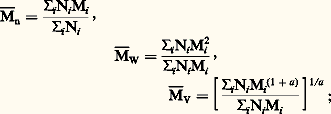 a est un coefficient qui dépend de la taille du polymère, de sa forme et de la qualité du solvant (cf. infra Viscosité intrinsèque ).Pour représenter la polymolécularité d’un système macromoléculaire, on peut, soit tracer la variation de la masse des espèces i d’un échantillon en fonction de leur masse molaire Mi , soit se satisfaire de son indice de polymolécularité,
a est un coefficient qui dépend de la taille du polymère, de sa forme et de la qualité du solvant (cf. infra Viscosité intrinsèque ).Pour représenter la polymolécularité d’un système macromoléculaire, on peut, soit tracer la variation de la masse des espèces i d’un échantillon en fonction de leur masse molaire Mi , soit se satisfaire de son indice de polymolécularité,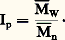 Au niveau microscopique, les macromolécules forment des ensembles dont la structure est désignée par le terme «morphologie». Les assemblages de pelotes statistiques donnent lieu à des enchevêtrements qui concourent à leur cohésion. Ces systèmes sont dits amorphes, car leur structure ne peut être décrite que par des lois statistiques et, au niveau macroscopique, la densité de matière est sensiblement constante pour tout élément de volume considéré.À l’opposé, pour des macromolécules particulièrement régulières, il est possible d’obtenir une cristallisation analogue à celle qui est observée chez les molécules simples et qui conduit à des monocristaux. Dans le cas des systèmes macromoléculaires, chaque chaîne donne lieu à des repliements réguliers qui répondent encore à une recherche de l’état énergétique minimal. La cristallisation des ploymères sous la forme de monocristaux est très difficile à obtenir, car elle implique le désenchevêtrement total des chaînes à partir de solutions; ces monocristaux restent donc des curiosités de laboratoire qui ne présentent, pour l’instant, qu’un intérêt théorique.Les morphologies intermédiaires entre ces deux extrêmes sont particulièrement intéressantes, puisqu’elles vont correspondre aux systèmes macromoléculaires présentant un certain niveau de régularité moléculaire. Ces systèmes très courants sont dits semi-cristallins et sont formés d’un assemblage de zones désordonnées (pelotes statistiques) et de zones plus ou moins cristallines. Pour caractériser cet état, on définit un taux de cristallinité X qui est égal à la fraction massique (Xm) ou volumique (Xv) des zones cristallines d’un échantillon donné. Le taux de cristallinité d’un polymère est un paramètre qui influence fortement ses propriétés physiques et mécaniques. En effet, la cohésion de la phase amorphe est très inférieure à celle des zones cristallines, et toutes les propriétés de l’état solide sont affectées par la variation de X. Toute propriété macroscopique spécifique 磊 pourra être mise sous la forme 磊= (1 — X) 磊a + 磊c, relation dans laquelle 磊a et 磊c désignent respectivement la propriété spécifique de la phase amorphe et celle de la phase cristalline.Pour les polymères à faible taux de cristallinité, la morphologie couramment admise est celle de micelles frangées, c’est-à-dire de structures vermiculaires alternant avec des séquences régulières qui peuvent s’associer pour donner des nodules cristallins, ou cristallites. Ces cristallites sont formées à partir de plusieurs chaînes, et toute chaîne peut participer à la formation de plusieurs cristallites. Elles sont dispersées dans une matrice amorphe et assurent une cohésion de l’ensemble par un phénomène de réticulation physique.Les polymères fortement cristallins font apparaître une organisation supérieure: ils s’arrangent en feuillets entre lesquels est rejetée la matière non cristallisable ou non cristallisée pour des raisons d’ordre cinétique. Au moyen de dislocations (défauts très organisés dans la structure), ces feuillets acquièrent une symétrie globale de type sphérique et les structures ainsi obtenues sont appelées sphérolites.La thermodynamique des systèmes macromoléculaires binaires permet d’expliquer la non-miscibilité des polymères entre eux. À quelques exceptions près, les macromolécules de nature différente ne se mélangent pas de façon homogène. En l’absence de tout additif, les matériaux macromoléculaires issus du mélange de deux polymères présentent des propriétés mécaniques médiocres. En revanche, l’incorporation à ces systèmes de copolymères à blocs ou greffé dont les blocs sont formés de chacun des polymères à mélanger et qui jouent le rôle d’émulgateurs, favorise la dispersion de la phase minoritaire dans la phase majoritaire et assure la cohésion à l’interphase. Sur le plan morphologique, cela conduit à des systèmes hétérogènes semblables à des émulsions solides dont les propriétés correspondent à une conjugaison de celles des deux homopolymères mélangés.2. Chimie macromoléculaireTrois aspects d’inégale importance sont rattachés au domaine de la chimie macromoléculaire. Le premier concerne la transformation chimique des polymères, naturels ou synthétiques, afin de modifier leurs propriétés et de les adapter à une application donnée. Le deuxième, beaucoup plus important eu égard à son potentiel et à ses applications, est relatif aux processus qui permettent de transformer un ensemble de molécules simples en chaînes macromoléculaires, et qu’on appelle polymérisations. Le troisième se rapporte aux transformations chimiques d’un composé macromoléculaire lorsqu’il est soumis à une agression chimique ou physique et qu’on appelle dégradations.Modification chimique des composés macromoléculairesC’est par modification chimique que les premiers polymères industriels, autres que naturels, ont été produits. En 1846, la nitration de la cellulose a conduit à un polymère artificiel (la nitrocellulose) dont les applications sont encore nombreuses de nos jours et qui, dès 1848, servait de base au Celluloïd (nom déposé d’une matière plastique).La plupart des modifications chimiques réalisées sur les polymères font intervenir des fonctions réactives situées latéralement par rapport au squelette macromoléculaire. La majeure partie des polymères industriels issus d’une modification chimique proviennent de l’estérification ou de l’éthérification de la cellulose:
Au niveau microscopique, les macromolécules forment des ensembles dont la structure est désignée par le terme «morphologie». Les assemblages de pelotes statistiques donnent lieu à des enchevêtrements qui concourent à leur cohésion. Ces systèmes sont dits amorphes, car leur structure ne peut être décrite que par des lois statistiques et, au niveau macroscopique, la densité de matière est sensiblement constante pour tout élément de volume considéré.À l’opposé, pour des macromolécules particulièrement régulières, il est possible d’obtenir une cristallisation analogue à celle qui est observée chez les molécules simples et qui conduit à des monocristaux. Dans le cas des systèmes macromoléculaires, chaque chaîne donne lieu à des repliements réguliers qui répondent encore à une recherche de l’état énergétique minimal. La cristallisation des ploymères sous la forme de monocristaux est très difficile à obtenir, car elle implique le désenchevêtrement total des chaînes à partir de solutions; ces monocristaux restent donc des curiosités de laboratoire qui ne présentent, pour l’instant, qu’un intérêt théorique.Les morphologies intermédiaires entre ces deux extrêmes sont particulièrement intéressantes, puisqu’elles vont correspondre aux systèmes macromoléculaires présentant un certain niveau de régularité moléculaire. Ces systèmes très courants sont dits semi-cristallins et sont formés d’un assemblage de zones désordonnées (pelotes statistiques) et de zones plus ou moins cristallines. Pour caractériser cet état, on définit un taux de cristallinité X qui est égal à la fraction massique (Xm) ou volumique (Xv) des zones cristallines d’un échantillon donné. Le taux de cristallinité d’un polymère est un paramètre qui influence fortement ses propriétés physiques et mécaniques. En effet, la cohésion de la phase amorphe est très inférieure à celle des zones cristallines, et toutes les propriétés de l’état solide sont affectées par la variation de X. Toute propriété macroscopique spécifique 磊 pourra être mise sous la forme 磊= (1 — X) 磊a + 磊c, relation dans laquelle 磊a et 磊c désignent respectivement la propriété spécifique de la phase amorphe et celle de la phase cristalline.Pour les polymères à faible taux de cristallinité, la morphologie couramment admise est celle de micelles frangées, c’est-à-dire de structures vermiculaires alternant avec des séquences régulières qui peuvent s’associer pour donner des nodules cristallins, ou cristallites. Ces cristallites sont formées à partir de plusieurs chaînes, et toute chaîne peut participer à la formation de plusieurs cristallites. Elles sont dispersées dans une matrice amorphe et assurent une cohésion de l’ensemble par un phénomène de réticulation physique.Les polymères fortement cristallins font apparaître une organisation supérieure: ils s’arrangent en feuillets entre lesquels est rejetée la matière non cristallisable ou non cristallisée pour des raisons d’ordre cinétique. Au moyen de dislocations (défauts très organisés dans la structure), ces feuillets acquièrent une symétrie globale de type sphérique et les structures ainsi obtenues sont appelées sphérolites.La thermodynamique des systèmes macromoléculaires binaires permet d’expliquer la non-miscibilité des polymères entre eux. À quelques exceptions près, les macromolécules de nature différente ne se mélangent pas de façon homogène. En l’absence de tout additif, les matériaux macromoléculaires issus du mélange de deux polymères présentent des propriétés mécaniques médiocres. En revanche, l’incorporation à ces systèmes de copolymères à blocs ou greffé dont les blocs sont formés de chacun des polymères à mélanger et qui jouent le rôle d’émulgateurs, favorise la dispersion de la phase minoritaire dans la phase majoritaire et assure la cohésion à l’interphase. Sur le plan morphologique, cela conduit à des systèmes hétérogènes semblables à des émulsions solides dont les propriétés correspondent à une conjugaison de celles des deux homopolymères mélangés.2. Chimie macromoléculaireTrois aspects d’inégale importance sont rattachés au domaine de la chimie macromoléculaire. Le premier concerne la transformation chimique des polymères, naturels ou synthétiques, afin de modifier leurs propriétés et de les adapter à une application donnée. Le deuxième, beaucoup plus important eu égard à son potentiel et à ses applications, est relatif aux processus qui permettent de transformer un ensemble de molécules simples en chaînes macromoléculaires, et qu’on appelle polymérisations. Le troisième se rapporte aux transformations chimiques d’un composé macromoléculaire lorsqu’il est soumis à une agression chimique ou physique et qu’on appelle dégradations.Modification chimique des composés macromoléculairesC’est par modification chimique que les premiers polymères industriels, autres que naturels, ont été produits. En 1846, la nitration de la cellulose a conduit à un polymère artificiel (la nitrocellulose) dont les applications sont encore nombreuses de nos jours et qui, dès 1848, servait de base au Celluloïd (nom déposé d’une matière plastique).La plupart des modifications chimiques réalisées sur les polymères font intervenir des fonctions réactives situées latéralement par rapport au squelette macromoléculaire. La majeure partie des polymères industriels issus d’une modification chimique proviennent de l’estérification ou de l’éthérification de la cellulose: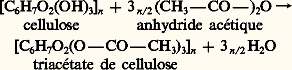 .Dans le domaine des polymères synthétiques, l’alcool polyvinylique ne peut être obtenu que par l’hydrolyse d’un poly(ester vinylique), car l’alcool vinylique monomère n’existe pas:
.Dans le domaine des polymères synthétiques, l’alcool polyvinylique ne peut être obtenu que par l’hydrolyse d’un poly(ester vinylique), car l’alcool vinylique monomère n’existe pas: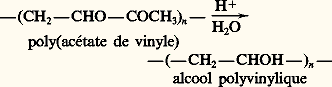 .Réactions de polymérisationIl existe deux grandes familles de polymérisations: les polymérisations par étapes (dont les polycondensations constituent la majorité) et les polymérisations en chaîne, qui ont acquis une importance majeure dans l’industrie chimique.Les polymérisations par étapesCes méthodes de polymérisation utilisent toutes les réactions de la chimie organique ou organométallique qui permettent de coupler deux ensembles moléculaires par l’intermédiaire d’une liaison covalente. Mais, pour que de telles réactions conduisent à des macromolécules, il est nécessaire, d’une part, que le nombre de liaisons générées (valence) à partir des molécules monomères soit au moins égal à deux et, d’autre part, que ces réactions soient totales ou que l’équilibre réactionnel puisse être déplacé. X et Y figurant les fonctions réactives antagonistes d’une molécule monomère, on peut les représenter par:
.Réactions de polymérisationIl existe deux grandes familles de polymérisations: les polymérisations par étapes (dont les polycondensations constituent la majorité) et les polymérisations en chaîne, qui ont acquis une importance majeure dans l’industrie chimique.Les polymérisations par étapesCes méthodes de polymérisation utilisent toutes les réactions de la chimie organique ou organométallique qui permettent de coupler deux ensembles moléculaires par l’intermédiaire d’une liaison covalente. Mais, pour que de telles réactions conduisent à des macromolécules, il est nécessaire, d’une part, que le nombre de liaisons générées (valence) à partir des molécules monomères soit au moins égal à deux et, d’autre part, que ces réactions soient totales ou que l’équilibre réactionnel puisse être déplacé. X et Y figurant les fonctions réactives antagonistes d’une molécule monomère, on peut les représenter par: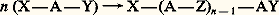 pour une réaction non équilibrée ou
pour une réaction non équilibrée ou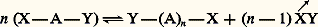 pour une polycondensation.Dans ces réactions, A représente le cœur de la molécule et Z le produit de réaction de X sur Y. Le schéma réactionnel de la formation du polyundécanamide (polyamide-11), qui est un polyamide thermoplastique semi-cristallin, peut s’écrire ainsi:
pour une polycondensation.Dans ces réactions, A représente le cœur de la molécule et Z le produit de réaction de X sur Y. Le schéma réactionnel de la formation du polyundécanamide (polyamide-11), qui est un polyamide thermoplastique semi-cristallin, peut s’écrire ainsi: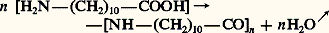 .Les polymérisations par étapes sont utilisées pour produire les polyesters, les polyamides, les polyuréthanes, les résines époxydiques, les phénoplastes et les aminoplastes ainsi que quelques familles de polymères de moindre importance économique.Les polymérisations en chaîneLes polymérisations en chaîne font généralement intervenir trois étapes essentielles. La première, appelée étape d’amorçage, correspond à l’activation d’une molécule monomère M par un centre actif primaire P généré par un amorceur A.
.Les polymérisations par étapes sont utilisées pour produire les polyesters, les polyamides, les polyuréthanes, les résines époxydiques, les phénoplastes et les aminoplastes ainsi que quelques familles de polymères de moindre importance économique.Les polymérisations en chaîneLes polymérisations en chaîne font généralement intervenir trois étapes essentielles. La première, appelée étape d’amorçage, correspond à l’activation d’une molécule monomère M par un centre actif primaire P généré par un amorceur A.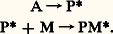 Dans un deuxième temps, le centre actif porté par la molécule monomère activée PM réagit à son tour avec une nouvelle molécule monomère M pour donner un nouveau centre, actif PMM, et ainsi de suite.
Dans un deuxième temps, le centre actif porté par la molécule monomère activée PM réagit à son tour avec une nouvelle molécule monomère M pour donner un nouveau centre, actif PMM, et ainsi de suite.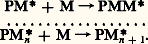 C’est au cours de cette étape, appelée propagation, qu’est formée la chaîne macromoléculaire. Le processus est généralement exothermique et son énergie d’activation positive; son contrôle nécessite donc l’intervention d’une réaction qui désactive les chaînes une fois formées; c’est l’étape de terminaison, schématisée par PMnPMn.Avec certains systèmes réactionnels, il est possible de générer plusieurs macromolécules à partir d’un seul centre actif primaire; ce processus est appelétransfert :
C’est au cours de cette étape, appelée propagation, qu’est formée la chaîne macromoléculaire. Le processus est généralement exothermique et son énergie d’activation positive; son contrôle nécessite donc l’intervention d’une réaction qui désactive les chaînes une fois formées; c’est l’étape de terminaison, schématisée par PMnPMn.Avec certains systèmes réactionnels, il est possible de générer plusieurs macromolécules à partir d’un seul centre actif primaire; ce processus est appelétransfert :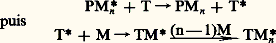 ,etc.Les réactions de polymérisation en chaîne peuvent être obtenues avec des molécules monomères de structure variée. Les plus courantes sont celles qui contiennent une double liaison éthylénique et dont la polymérisation correspond à la transformation de la liaison 神 en liaison 靖.
,etc.Les réactions de polymérisation en chaîne peuvent être obtenues avec des molécules monomères de structure variée. Les plus courantes sont celles qui contiennent une double liaison éthylénique et dont la polymérisation correspond à la transformation de la liaison 神 en liaison 靖.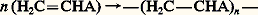 Selon le même principe, les groupes
Selon le même principe, les groupes peuvent être polymérisés pour donner respectivement
peuvent être polymérisés pour donner respectivement Les hétérocycles se polymérisent par l’ouverture des cycles
Les hétérocycles se polymérisent par l’ouverture des cycles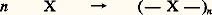 X correspondant à l’hétéroélément responsable de la polymérisabilité du monomère.Le classement des méthodes de polymérisation en chaîne est généralement fait à partir des différents types de centres actifs responsables du processus de polymérisation. On distingue ainsi quatre types de polymérisations en chaîne.La polymérisation radicalaire est propagée par des radicaux libres générés par des amorceurs lors d’un processus physique (thermique, photochimique...) ou chimique (rupture homolytique d’une liaison 靖, réaction d’oxydo-réduction...). Ces radicaux libres donnent lieu à des additions radicalaires sur les doubles liaisons carbone-carbone. Les amorceurs les plus couramment utilisés sont ceux qui engendrent le centre actif primaire P, par rupture homolytique d’une liaison covalente : peroxydes organiques ou minéraux (rupture de la liaison —O—O—), diazoïques...La durée de vie d’une chaîne en croissance est extrêmement courte (de l’ordre de la seconde). La terminaison de la chaîne fait intervenir la rencontre de deux radicaux libres portés par deux chaînes en croissance. Les différentes étapes de la polymérisation radicalaire du styrène amorcée par le peroxyde de benzoyle sont les suivantes:
X correspondant à l’hétéroélément responsable de la polymérisabilité du monomère.Le classement des méthodes de polymérisation en chaîne est généralement fait à partir des différents types de centres actifs responsables du processus de polymérisation. On distingue ainsi quatre types de polymérisations en chaîne.La polymérisation radicalaire est propagée par des radicaux libres générés par des amorceurs lors d’un processus physique (thermique, photochimique...) ou chimique (rupture homolytique d’une liaison 靖, réaction d’oxydo-réduction...). Ces radicaux libres donnent lieu à des additions radicalaires sur les doubles liaisons carbone-carbone. Les amorceurs les plus couramment utilisés sont ceux qui engendrent le centre actif primaire P, par rupture homolytique d’une liaison covalente : peroxydes organiques ou minéraux (rupture de la liaison —O—O—), diazoïques...La durée de vie d’une chaîne en croissance est extrêmement courte (de l’ordre de la seconde). La terminaison de la chaîne fait intervenir la rencontre de deux radicaux libres portés par deux chaînes en croissance. Les différentes étapes de la polymérisation radicalaire du styrène amorcée par le peroxyde de benzoyle sont les suivantes: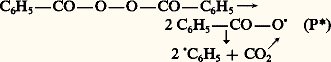
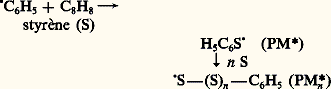
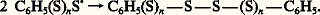 .La polymérisation radicalaire est utilisée pour polymériser l’éthylène ainsi que la plupart des monomères vinyliques (chlorure de vinyle, acétate de vinyle, styrène...) et assimilés (monomères acryliques et méthacryliques, tétrafluoroéthylène, diènes...). Cette méthode de polymérisation est particulièrement bien adaptée à la variété des techniques (masse, émulsion, solution, suspension) couramment utilisées pour la production des polymères.Lorsque le centre actif est polarisé (ou chargé) négativement, il peut donner lieu à des réactions nucléophiles et les polymérisations correspondantes sont appelées polymérisations anioniques . Les amorceurs les plus sollicités pour de telles réactions sont des composés organométalliques, tel le butyllithium, qui sont des bases de Lewis (entités qui peuvent donner des électrons) extrêmement fortes. Avec des monomères éthyléniques (styrène, méthacrylate de méthyle...) ou diéniques (butadiène, isoprène), la polymérisation est obtenue par addition nucléophile sur la double liaison. Cette dernière est activée par des substituants qui permettent sa polarisation positive au moment de l’attaque du centre actif. Dans le cas des hétérocycles, la réaction de base peut être une substitution nucléophile (polymérisation des oxirannes), une addition-élimination sur un carbonyle (cas des lactones) ou toute autre réaction susceptible d’ouvrir le cycle.Les polymérisations cationiques mettent en œuvre les processus symétriques des précédents. Les centres actifs propageants sont des espèces fortement électrophiles, chargées (ou polarisées) positivement, et ils sont générés par des acides protoniques ou des acides de Lewis (entités qui acceptent des électrons). Dans ce cas et pour des monomères de type éthylénique, la polymérisabilité est accrue par les substituants de la double liaison qui ont un effet électrodonneur [par exemple, il en est ainsi pour l’isobutène, (CH3)2C=CH2] ou par les substituants qui peuvent autoriser une polarisation négative de la double liaison au moment de l’approche du centre actif. Les monomères hétérocycliques qui se polymérisent selon un processus cationique présentent des sites nucléophiles capables de donner une réaction acide-base avec le centre actif propageant.La polymérisation par coordination recouvre une grande variété de systèmes qui ont en commun la présence d’un atome d’un métal de transition à l’extrémité de la chaîne en croissance. Les monomères (éthyléniques ou hétérocycliques) possèdent un caractère basique au sens de Lewis, qui va permettre leur coordination (fixation) par l’intermédiaire des orbitales vacantes du métal de transition. L’effet de cette coordination est double: fixation de la molécule monomère dans une géométrie déterminée par rapport à la chaîne en croissance et affaiblissement simultané des liaisons fragiles du monomère. Le premier effet permet d’obtenir des polymérisations stéréospécifiques (polypropène isotactique ou syndiotactique), le second explique la très grande polymérisabilité des monomères oléfiniques en présence de certains complexes de coordination et la régularité structurale des polymères obtenus (polyéthylène linéaire).Les innovations actuellement développées à partir des polymérisations en chaîne visent à obtenir un meilleur contrôle des processus. Il peut s’agir d’accroître ou de diversifier les régularités configurationnelles des macromolécules synthétisées ou bien de faire en sorte que les étapes habituelles de terminaison ou de transfert soient négligeables ou supprimées. Dans le premier cas, on qualifie les polymérisations correspondantes de contrôlées, alors que dans le second on parle de polymérisations vivantes. Lorsque la durée de l’étape d’amorçage est faible par rapport à celle de la propagation, il est possible d’obtenir des polymères à faible indice de polymolécularité et d’accéder à des architectures particulières. Les propriétés des matériaux qui en découlent sont différentes de celles des polymères traditionnels et permettent un élargissement du champ d’application des composés macromoléculaires.Jusqu’à une date récente, seules les polymérisations anioniques pouvaient prétendre au caractère vivant. Certaines polymérisations cationiques ou par coordination entrent à présent dans cette catégorie; les polymérisations radicalaires se prêtent aussi, depuis peu de temps, à un certain contrôle de l’étape de propagation.Les techniques mises en œuvre pour réaliser les polymérisations peuvent être classées en quatre catégories. Les polymérisations en masse , dans lesquelles l’essentiel de la masse du système réactionnel est constitué par le monomère et par le polymère en cours de formation, sont couramment utilisées pour les polymérisations par étapes. Pour les polymérisations en chaîne, l’accroissement de la viscosité du milieu au fur et à mesure de la conversion du monomère en polymère rend délicat le contrôle du processus, lequel est généralement exothermique. Pour pallier cela, il est possible d’opérer en solution dans un solvant du polymère, en suspension dans une phase dispersante aqueuse ou en émulsion (en présence d’agents tensioactifs). Les peintures aqueuses (vinyliques ou acryliques) sont obtenues à partir de cette dernière technique.Dégradation des composés macromoléculairesLa dégradation est l’une des limitations majeures de l’utilisation des polymères comme matériaux. En effet, le lien étroit entre les caractéristiques mécaniques et la structure moléculaire (en particulier les dimensions moléculaires) conduit à n’utiliser les matériaux macromoléculaires que dans des environnements physiques (ou parfois chimiques) peu agressifs. Les réactions de dégradation peuvent résulter d’une action physique (chaleur, radiation électromagnétique, radiation ionisante) ou chimique (hydrolyse, oxydation). Les effets les plus spectaculaires sont observés quand les deux types d’agressions sont combinés (photo-oxydation, thermo-oxydation...).Les dégradations procèdent selon deux schémas réactionnels principaux: par la rupture aléatoire des liaisons covalentes qui assurent le lien entre les motifs constitutifs ou par la dépolymérisation thermique de la chaîne à une température supérieure à la température plafond du monomère correspondant. Dans ce dernier cas et pour des raisons thermodynamiques, le monomère est régénéré par le processus inverse de la polymérisation en chaîne.Les tendances actuelles de la recherche en chimie macromoléculaire privilégient l’amélioration des procédés existants et la diversification des architectures macromoléculaires plutôt que la préparation de macromolécules à partir de nouveaux monomères, de structure forcément plus complexe.3. Étude des macromolécules en solutionObjectifsDepuis son origine, la science des polymères est confrontée à la problématique de la détermination de la taille des macromolécules et de la forme qu’elles adoptent dans l’espace. La connaissance de la conformation des chaînes dans un environnement donné ainsi que la corrélation de cette information avec les caractéristiques structurales de l’échantillon considéré (nature chimique du motif constitutif, masse molaire moyenne, distribution des masses) constituent un préalable à toute étude sur son comportement physico-chimique ou physico-mécanique. Si la masse molaire moyenne d’un échantillon et sa polymolécularité dépendent avant tout des conditions de synthèse choisies par l’expérimentateur, la conformation des chaînes de polymères résulte, quant à elle, de l’interrelation entre la structure de leurs unités constitutives et le milieu physique dans lequel ces chaînes sont placées (présence de solvant ou d’autres polymères, température, état étiré ou au repos). Indépendamment de ces considérations, il apparaît que c’est en solution (à l’état de soluté) qu’un échantillon peut être le plus aisément caractérisé (à l’exception notable de la technique de diffusion neutronique, qui ne nécessite pas la présence d’un solvant pour permettre la caractérisation du polymère étudié).L’osmométrie, la diffusion de la lumière, la chromatographie d’exclusion stérique ou la viscosimétrie, techniques qui requièrent toutes des solutions diluées de polymères, sont des méthodes de caractérisation très utilisées et complémentaires. Elles permettent d’accéder à des informations aussi diverses que la polymolécularité, les masses molaires moyennes, le rayon de giration ou la forme des macromolécules.Outre la détermination des caractéristiques moléculaires d’un échantillon, l’étude des solutions macromoléculaires a permis de connaître les facteurs qui influent sur le comportement des polymères en solution. Le nombre d’applications faisant intervenir des polymères en solution (peintures, produits cosmétiques ou alimentaires, etc.) atteste le remarquable effort cognitif dont ce domaine a bénéficié.Facteurs affectant la conformation des chaînesUne solution contenant des macromolécules diffère d’une solution de molécules organiques simples en premier lieu par la répercussion à longue distance des interactions qui peuvent s’y développer. La grande taille et la connectivité (c’est-à-dire le fait que toutes les unités monomères de la chaîne soient liées les unes aux autres) d’une chaîne macromoléculaire impliquent, en effet, qu’elle peut interagir avec d’autres éléments de la solution sur plusieurs dizaines voire plusieurs centaines de nanomètres et cela de multiples manières. Ces interactions sont, par exemple, celles qui s’établissent entre les motifs constitutifs de la chaîne et les molécules de solvant, interactions qui, répétées de nombreuses fois, contribuent à allonger la chaîne; les interactions peuvent aussi concerner le contact temporaire entre deux chaînes de la solution à travers la formation d’un enchevêtrement.Modèle de la marche au hasardPour comprendre les fondements du comportement d’une macromolécule en solution, il faut tout d’abord connaître chacun des facteurs qui concourent à lui donner une certaine dimension. Le modèle qui décrit le mieux la conformation d’une macromolécule est celui de la marche au hasard, dans lequel les chaînes sont considérées comme immatérielles ou fantômes. Ce modèle permet de calculer, entre autres grandeurs, la distance r qui sépare les deux extrémités de la chaîne polymère, distance qui donne une idée de la taille que prend la macromolécule. Ce vecteur r est, en réalité, la somme des vecteurs correspondant à la longueur (L ) de chacune des N unités de répétition:
.La polymérisation radicalaire est utilisée pour polymériser l’éthylène ainsi que la plupart des monomères vinyliques (chlorure de vinyle, acétate de vinyle, styrène...) et assimilés (monomères acryliques et méthacryliques, tétrafluoroéthylène, diènes...). Cette méthode de polymérisation est particulièrement bien adaptée à la variété des techniques (masse, émulsion, solution, suspension) couramment utilisées pour la production des polymères.Lorsque le centre actif est polarisé (ou chargé) négativement, il peut donner lieu à des réactions nucléophiles et les polymérisations correspondantes sont appelées polymérisations anioniques . Les amorceurs les plus sollicités pour de telles réactions sont des composés organométalliques, tel le butyllithium, qui sont des bases de Lewis (entités qui peuvent donner des électrons) extrêmement fortes. Avec des monomères éthyléniques (styrène, méthacrylate de méthyle...) ou diéniques (butadiène, isoprène), la polymérisation est obtenue par addition nucléophile sur la double liaison. Cette dernière est activée par des substituants qui permettent sa polarisation positive au moment de l’attaque du centre actif. Dans le cas des hétérocycles, la réaction de base peut être une substitution nucléophile (polymérisation des oxirannes), une addition-élimination sur un carbonyle (cas des lactones) ou toute autre réaction susceptible d’ouvrir le cycle.Les polymérisations cationiques mettent en œuvre les processus symétriques des précédents. Les centres actifs propageants sont des espèces fortement électrophiles, chargées (ou polarisées) positivement, et ils sont générés par des acides protoniques ou des acides de Lewis (entités qui acceptent des électrons). Dans ce cas et pour des monomères de type éthylénique, la polymérisabilité est accrue par les substituants de la double liaison qui ont un effet électrodonneur [par exemple, il en est ainsi pour l’isobutène, (CH3)2C=CH2] ou par les substituants qui peuvent autoriser une polarisation négative de la double liaison au moment de l’approche du centre actif. Les monomères hétérocycliques qui se polymérisent selon un processus cationique présentent des sites nucléophiles capables de donner une réaction acide-base avec le centre actif propageant.La polymérisation par coordination recouvre une grande variété de systèmes qui ont en commun la présence d’un atome d’un métal de transition à l’extrémité de la chaîne en croissance. Les monomères (éthyléniques ou hétérocycliques) possèdent un caractère basique au sens de Lewis, qui va permettre leur coordination (fixation) par l’intermédiaire des orbitales vacantes du métal de transition. L’effet de cette coordination est double: fixation de la molécule monomère dans une géométrie déterminée par rapport à la chaîne en croissance et affaiblissement simultané des liaisons fragiles du monomère. Le premier effet permet d’obtenir des polymérisations stéréospécifiques (polypropène isotactique ou syndiotactique), le second explique la très grande polymérisabilité des monomères oléfiniques en présence de certains complexes de coordination et la régularité structurale des polymères obtenus (polyéthylène linéaire).Les innovations actuellement développées à partir des polymérisations en chaîne visent à obtenir un meilleur contrôle des processus. Il peut s’agir d’accroître ou de diversifier les régularités configurationnelles des macromolécules synthétisées ou bien de faire en sorte que les étapes habituelles de terminaison ou de transfert soient négligeables ou supprimées. Dans le premier cas, on qualifie les polymérisations correspondantes de contrôlées, alors que dans le second on parle de polymérisations vivantes. Lorsque la durée de l’étape d’amorçage est faible par rapport à celle de la propagation, il est possible d’obtenir des polymères à faible indice de polymolécularité et d’accéder à des architectures particulières. Les propriétés des matériaux qui en découlent sont différentes de celles des polymères traditionnels et permettent un élargissement du champ d’application des composés macromoléculaires.Jusqu’à une date récente, seules les polymérisations anioniques pouvaient prétendre au caractère vivant. Certaines polymérisations cationiques ou par coordination entrent à présent dans cette catégorie; les polymérisations radicalaires se prêtent aussi, depuis peu de temps, à un certain contrôle de l’étape de propagation.Les techniques mises en œuvre pour réaliser les polymérisations peuvent être classées en quatre catégories. Les polymérisations en masse , dans lesquelles l’essentiel de la masse du système réactionnel est constitué par le monomère et par le polymère en cours de formation, sont couramment utilisées pour les polymérisations par étapes. Pour les polymérisations en chaîne, l’accroissement de la viscosité du milieu au fur et à mesure de la conversion du monomère en polymère rend délicat le contrôle du processus, lequel est généralement exothermique. Pour pallier cela, il est possible d’opérer en solution dans un solvant du polymère, en suspension dans une phase dispersante aqueuse ou en émulsion (en présence d’agents tensioactifs). Les peintures aqueuses (vinyliques ou acryliques) sont obtenues à partir de cette dernière technique.Dégradation des composés macromoléculairesLa dégradation est l’une des limitations majeures de l’utilisation des polymères comme matériaux. En effet, le lien étroit entre les caractéristiques mécaniques et la structure moléculaire (en particulier les dimensions moléculaires) conduit à n’utiliser les matériaux macromoléculaires que dans des environnements physiques (ou parfois chimiques) peu agressifs. Les réactions de dégradation peuvent résulter d’une action physique (chaleur, radiation électromagnétique, radiation ionisante) ou chimique (hydrolyse, oxydation). Les effets les plus spectaculaires sont observés quand les deux types d’agressions sont combinés (photo-oxydation, thermo-oxydation...).Les dégradations procèdent selon deux schémas réactionnels principaux: par la rupture aléatoire des liaisons covalentes qui assurent le lien entre les motifs constitutifs ou par la dépolymérisation thermique de la chaîne à une température supérieure à la température plafond du monomère correspondant. Dans ce dernier cas et pour des raisons thermodynamiques, le monomère est régénéré par le processus inverse de la polymérisation en chaîne.Les tendances actuelles de la recherche en chimie macromoléculaire privilégient l’amélioration des procédés existants et la diversification des architectures macromoléculaires plutôt que la préparation de macromolécules à partir de nouveaux monomères, de structure forcément plus complexe.3. Étude des macromolécules en solutionObjectifsDepuis son origine, la science des polymères est confrontée à la problématique de la détermination de la taille des macromolécules et de la forme qu’elles adoptent dans l’espace. La connaissance de la conformation des chaînes dans un environnement donné ainsi que la corrélation de cette information avec les caractéristiques structurales de l’échantillon considéré (nature chimique du motif constitutif, masse molaire moyenne, distribution des masses) constituent un préalable à toute étude sur son comportement physico-chimique ou physico-mécanique. Si la masse molaire moyenne d’un échantillon et sa polymolécularité dépendent avant tout des conditions de synthèse choisies par l’expérimentateur, la conformation des chaînes de polymères résulte, quant à elle, de l’interrelation entre la structure de leurs unités constitutives et le milieu physique dans lequel ces chaînes sont placées (présence de solvant ou d’autres polymères, température, état étiré ou au repos). Indépendamment de ces considérations, il apparaît que c’est en solution (à l’état de soluté) qu’un échantillon peut être le plus aisément caractérisé (à l’exception notable de la technique de diffusion neutronique, qui ne nécessite pas la présence d’un solvant pour permettre la caractérisation du polymère étudié).L’osmométrie, la diffusion de la lumière, la chromatographie d’exclusion stérique ou la viscosimétrie, techniques qui requièrent toutes des solutions diluées de polymères, sont des méthodes de caractérisation très utilisées et complémentaires. Elles permettent d’accéder à des informations aussi diverses que la polymolécularité, les masses molaires moyennes, le rayon de giration ou la forme des macromolécules.Outre la détermination des caractéristiques moléculaires d’un échantillon, l’étude des solutions macromoléculaires a permis de connaître les facteurs qui influent sur le comportement des polymères en solution. Le nombre d’applications faisant intervenir des polymères en solution (peintures, produits cosmétiques ou alimentaires, etc.) atteste le remarquable effort cognitif dont ce domaine a bénéficié.Facteurs affectant la conformation des chaînesUne solution contenant des macromolécules diffère d’une solution de molécules organiques simples en premier lieu par la répercussion à longue distance des interactions qui peuvent s’y développer. La grande taille et la connectivité (c’est-à-dire le fait que toutes les unités monomères de la chaîne soient liées les unes aux autres) d’une chaîne macromoléculaire impliquent, en effet, qu’elle peut interagir avec d’autres éléments de la solution sur plusieurs dizaines voire plusieurs centaines de nanomètres et cela de multiples manières. Ces interactions sont, par exemple, celles qui s’établissent entre les motifs constitutifs de la chaîne et les molécules de solvant, interactions qui, répétées de nombreuses fois, contribuent à allonger la chaîne; les interactions peuvent aussi concerner le contact temporaire entre deux chaînes de la solution à travers la formation d’un enchevêtrement.Modèle de la marche au hasardPour comprendre les fondements du comportement d’une macromolécule en solution, il faut tout d’abord connaître chacun des facteurs qui concourent à lui donner une certaine dimension. Le modèle qui décrit le mieux la conformation d’une macromolécule est celui de la marche au hasard, dans lequel les chaînes sont considérées comme immatérielles ou fantômes. Ce modèle permet de calculer, entre autres grandeurs, la distance r qui sépare les deux extrémités de la chaîne polymère, distance qui donne une idée de la taille que prend la macromolécule. Ce vecteur r est, en réalité, la somme des vecteurs correspondant à la longueur (L ) de chacune des N unités de répétition: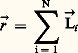 En raison de l’agitation thermique, la mesure de la valeur instantanée de cette grandeur n’est pas envisageable. Quant à sa valeur moyenne, obtenue sur une longue période de temps, elle serait égale à 0. Il est donc plus pratique de calculer la valeur quadratique du vecteur r et de l’exprimer sous la forme d’une moyenne:
En raison de l’agitation thermique, la mesure de la valeur instantanée de cette grandeur n’est pas envisageable. Quant à sa valeur moyenne, obtenue sur une longue période de temps, elle serait égale à 0. Il est donc plus pratique de calculer la valeur quadratique du vecteur r et de l’exprimer sous la forme d’une moyenne: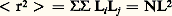 麗礪,Dans ce calcul de 麗r2 礪, ou écart quadratique moyen, S. Chandrasekhar n’a pas pris en compte les contraintes locales que peuvent subir les segments des chaînes macromoléculaires. Les chaînes sont en effet soumises à deux types de restrictions que le modèle de la marche au hasard ignore totalement: les angles de valence entre atomes sont fixes et l’encombrement stérique empêche la libre rotation de la chaîne autour de ses segments. H. Benoit a donc proposé une relation qui tient compte de ces interactions locales ayant pour effet d’étirer les chaînes:
麗礪,Dans ce calcul de 麗r2 礪, ou écart quadratique moyen, S. Chandrasekhar n’a pas pris en compte les contraintes locales que peuvent subir les segments des chaînes macromoléculaires. Les chaînes sont en effet soumises à deux types de restrictions que le modèle de la marche au hasard ignore totalement: les angles de valence entre atomes sont fixes et l’encombrement stérique empêche la libre rotation de la chaîne autour de ses segments. H. Benoit a donc proposé une relation qui tient compte de ces interactions locales ayant pour effet d’étirer les chaînes: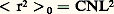 麗礪.C est un facteur qui varie en fonction de la rigidité de la chaîne; il peut prendre des valeurs entre 4 (chaîne flexible) et 10 (chaîne rigide). Quant à la distribution des valeurs que peut prendre S20, elle obéit à une courbe de distribution gaussienne. À partir de l’expression de 麗r2 礪0, il est possible de définir d’autres grandeurs, tel le rayon de giration:
麗礪.C est un facteur qui varie en fonction de la rigidité de la chaîne; il peut prendre des valeurs entre 4 (chaîne flexible) et 10 (chaîne rigide). Quant à la distribution des valeurs que peut prendre S20, elle obéit à une courbe de distribution gaussienne. À partir de l’expression de 麗r2 礪0, il est possible de définir d’autres grandeurs, tel le rayon de giration: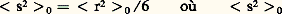 麗礪麗礪麗礪est le carré moyen de la distance d’un segment quelconque de la chaîne à son centre de masse.Interactions à longue distance et volume excluJusqu’ici, seules les interactions à courte distance, propres à la chaîne, ont été prises en compte. Mais plongés dans un bon solvant, les segments de la chaîne tendent à minimiser leurs contacts et à s’entourer de molécules de solvant, contribuant à créer des interactions à longue distance. Ce phénomène dit «de volume exclu», qui empêche deux éléments de la chaîne de se recouvrir, aboutit au gonflement de la chaîne: le modèle de la marche au hasard n’est donc plus valide. Deux situations particulières correspondent, cependant, au cas idéal de la chaîne gaussienne non perturbée que prévoit ce modèle. En présence d’un solvant intermédiaire sur le plan thermodynamique, entre le bon solvant et le précipitant, à une température (T size=1) située un peu au-dessus du point de précipitation, la tendance des éléments de la chaîne à se repousser est exactement compensée par leur tendance à s’attirer. Dans cette situation, nous sommes dans des conditions dites «thêta» (la chaîne ne gonfle pas). Le second cas de figure correspond à celui d’un polymère à l’état fondu: les chaînes se comportent là aussi de façon idéale et gaussienne, la tendance d’un élément de chaîne à repousser le chaînon adjacent étant annulée par l’interaction avec un élément d’une autre chaîne.Pour revenir au cas du polymère dissous dans un bon solvant, l’expansion peut être évaluée en égalant les deux termes énergétiques: l’un, d’origine osmotique (gonflement), exercé par le solvant, et l’autre, d’origine élastique, correspondant à la résistance de la chaîne. Dans ces conditions, l’écart quadratique moyen s’écrit:
麗礪麗礪麗礪est le carré moyen de la distance d’un segment quelconque de la chaîne à son centre de masse.Interactions à longue distance et volume excluJusqu’ici, seules les interactions à courte distance, propres à la chaîne, ont été prises en compte. Mais plongés dans un bon solvant, les segments de la chaîne tendent à minimiser leurs contacts et à s’entourer de molécules de solvant, contribuant à créer des interactions à longue distance. Ce phénomène dit «de volume exclu», qui empêche deux éléments de la chaîne de se recouvrir, aboutit au gonflement de la chaîne: le modèle de la marche au hasard n’est donc plus valide. Deux situations particulières correspondent, cependant, au cas idéal de la chaîne gaussienne non perturbée que prévoit ce modèle. En présence d’un solvant intermédiaire sur le plan thermodynamique, entre le bon solvant et le précipitant, à une température (T size=1) située un peu au-dessus du point de précipitation, la tendance des éléments de la chaîne à se repousser est exactement compensée par leur tendance à s’attirer. Dans cette situation, nous sommes dans des conditions dites «thêta» (la chaîne ne gonfle pas). Le second cas de figure correspond à celui d’un polymère à l’état fondu: les chaînes se comportent là aussi de façon idéale et gaussienne, la tendance d’un élément de chaîne à repousser le chaînon adjacent étant annulée par l’interaction avec un élément d’une autre chaîne.Pour revenir au cas du polymère dissous dans un bon solvant, l’expansion peut être évaluée en égalant les deux termes énergétiques: l’un, d’origine osmotique (gonflement), exercé par le solvant, et l’autre, d’origine élastique, correspondant à la résistance de la chaîne. Dans ces conditions, l’écart quadratique moyen s’écrit: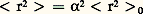 麗礪麗礪où 見 est le facteur d’expansion, qui correspond approximativement à 1/10, ce qui donne, pour une chaîne comportant 1 000 unités monomères (1 000 N), un accroissement de ses dimensions d’un facteur 2 et de son volume d’un facteur 8. C’est en ces termes que Paul John Flory a traité le problème du volume exclu, à partir d’une théorie de champ moyen qui fait l’hypothèse d’une concentration uniforme en segments de polymères dans la solution. Même si elle ne reflète que partiellement la réalité physique (la concentration est sujette à des fluctuations importantes en milieu dilué), cette description a fait l’objet de vérifications expérimentales dans un certain nombre de cas.Trois régimes de concentrationLes travaux de P.-G de Gennes et ses collaborateurs sont venus compléter la théorie de Flory, qui s’est montrée impuissante à rendre compte de la variation des dimensions d’une chaîne avec la concentration du milieu. Tant qu’on se situe dans le domaine des solutions diluées, la grandeur caractéristique à considérer est l’une des deux dimensions définies auparavant,
麗礪麗礪où 見 est le facteur d’expansion, qui correspond approximativement à 1/10, ce qui donne, pour une chaîne comportant 1 000 unités monomères (1 000 N), un accroissement de ses dimensions d’un facteur 2 et de son volume d’un facteur 8. C’est en ces termes que Paul John Flory a traité le problème du volume exclu, à partir d’une théorie de champ moyen qui fait l’hypothèse d’une concentration uniforme en segments de polymères dans la solution. Même si elle ne reflète que partiellement la réalité physique (la concentration est sujette à des fluctuations importantes en milieu dilué), cette description a fait l’objet de vérifications expérimentales dans un certain nombre de cas.Trois régimes de concentrationLes travaux de P.-G de Gennes et ses collaborateurs sont venus compléter la théorie de Flory, qui s’est montrée impuissante à rendre compte de la variation des dimensions d’une chaîne avec la concentration du milieu. Tant qu’on se situe dans le domaine des solutions diluées, la grandeur caractéristique à considérer est l’une des deux dimensions définies auparavant,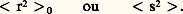 麗礪麗礪.À mesure que la concentration augmente, on atteint un régime dit semi-dilué, dans lequel les chaînes commencent à se recouvrir (interagir entre elles). Ce phénomène intervient à partir d’une certaine concentration, appelée concentration critique de recouvrement (C). Au-delà de C, 麗s2 礪0 perd progressivement son sens comme grandeur contrôlant le comportement des solutions de polymères. La grandeur pertinente est dorénavant 﨡, qui caractérise la distance moyenne qui sépare deux points de contact d’une même chaîne avec deux chaînes étrangères. Cette distance décroît à mesure que la concentration augmente. C’est ainsi que pour C 礪 C, l’effet de volume exclu pour une chaîne est restreint aux distances inférieures à 﨡. La chaîne peut être représentée comme une chaîne gaussienne constituée d’éléments de taille 﨡 et ses dimensions caractéristiques sont données par le rayon de giration s( 﨏) qui est proportionnel à s ( 﨏) size=1憐 﨡2 ( 﨏).s( 﨏) dépend de la concentration et représente le nombre de motifs contitutifs encore soumis à l’effet de volume exclu et ( 﨏) représente la fraction volumique du polymère. On démontre ainsi que le rapport des rayons de giration à l’état semi-dilué (C 礪 C) et à C est égal à:
麗礪麗礪.À mesure que la concentration augmente, on atteint un régime dit semi-dilué, dans lequel les chaînes commencent à se recouvrir (interagir entre elles). Ce phénomène intervient à partir d’une certaine concentration, appelée concentration critique de recouvrement (C). Au-delà de C, 麗s2 礪0 perd progressivement son sens comme grandeur contrôlant le comportement des solutions de polymères. La grandeur pertinente est dorénavant 﨡, qui caractérise la distance moyenne qui sépare deux points de contact d’une même chaîne avec deux chaînes étrangères. Cette distance décroît à mesure que la concentration augmente. C’est ainsi que pour C 礪 C, l’effet de volume exclu pour une chaîne est restreint aux distances inférieures à 﨡. La chaîne peut être représentée comme une chaîne gaussienne constituée d’éléments de taille 﨡 et ses dimensions caractéristiques sont données par le rayon de giration s( 﨏) qui est proportionnel à s ( 﨏) size=1憐 﨡2 ( 﨏).s( 﨏) dépend de la concentration et représente le nombre de motifs contitutifs encore soumis à l’effet de volume exclu et ( 﨏) représente la fraction volumique du polymère. On démontre ainsi que le rapport des rayons de giration à l’état semi-dilué (C 礪 C) et à C est égal à: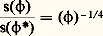 .Dans ce cas, la chaîne se contracte progressivement jusqu’à retrouver ses dimensions non perturbées en milieu concentré (absence de volume exclu).Méthodes de détermination des masses molairesThermodynamique des solutionsNotre compréhension du comportement en solution des polymères ne serait pas totale si nous ne prenions en compte que les restrictions qui viennent d’être décrites. La qualité du solvant utilisé et l’affinité qu’il a pour la chaîne polymère, sont le reflet de la compétition entre les interactions segment-segment et les interactions segment-solvant. Ces caractéristiques du solvant affectent, autant que les facteurs précédemment cités, la conformation qu’adopte la chaîne macromoléculaire. À cette différence près que le volume molaire du soluté est très supérieur à celui du solvant, la description de ces interactions s’inscrit dans le cadre de la thermodynamique des solutions qui, de plus, stipule que la miscibilité de deux constituants est conditionnée par une énergie libre de mélange négative:
.Dans ce cas, la chaîne se contracte progressivement jusqu’à retrouver ses dimensions non perturbées en milieu concentré (absence de volume exclu).Méthodes de détermination des masses molairesThermodynamique des solutionsNotre compréhension du comportement en solution des polymères ne serait pas totale si nous ne prenions en compte que les restrictions qui viennent d’être décrites. La qualité du solvant utilisé et l’affinité qu’il a pour la chaîne polymère, sont le reflet de la compétition entre les interactions segment-segment et les interactions segment-solvant. Ces caractéristiques du solvant affectent, autant que les facteurs précédemment cités, la conformation qu’adopte la chaîne macromoléculaire. À cette différence près que le volume molaire du soluté est très supérieur à celui du solvant, la description de ces interactions s’inscrit dans le cadre de la thermodynamique des solutions qui, de plus, stipule que la miscibilité de deux constituants est conditionnée par une énergie libre de mélange négative: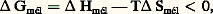 麗où Hmél est l’enthalpie du mélange, Smél l’entropie du mélange et T la température.La théorie de Flory-Huggins constitue la première tentative de conceptualisation d’un mélange constitué d’une macromolécule et de petites molécules de solvants. Cette théorie, qui a été élaborée sur le même modèle que celui des solutions régulières (formule ci-dessus), identifie la solution à un maillage dont les sites seraient occupés soit par des molécules de solvant, soit par un segment de chaîne.Le volume de ce réseau est considéré comme étant incompressible (toutes les mailles sont occupées), et la concentration du polymère est uniforme dans toute la solution, deux hypothèses qui ont quelque peu limité la portée de la théorie de Flory-Huggins, comme nous le verrons ultérieurement.L’expression de l’énergie libre de mélange par site du réseau dans un mélange solvant-polymère s’écrit:
麗où Hmél est l’enthalpie du mélange, Smél l’entropie du mélange et T la température.La théorie de Flory-Huggins constitue la première tentative de conceptualisation d’un mélange constitué d’une macromolécule et de petites molécules de solvants. Cette théorie, qui a été élaborée sur le même modèle que celui des solutions régulières (formule ci-dessus), identifie la solution à un maillage dont les sites seraient occupés soit par des molécules de solvant, soit par un segment de chaîne.Le volume de ce réseau est considéré comme étant incompressible (toutes les mailles sont occupées), et la concentration du polymère est uniforme dans toute la solution, deux hypothèses qui ont quelque peu limité la portée de la théorie de Flory-Huggins, comme nous le verrons ultérieurement.L’expression de l’énergie libre de mélange par site du réseau dans un mélange solvant-polymère s’écrit: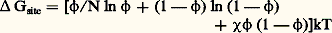 où k est la constante de Boltzmann.Les deux premiers termes de cette formule sont des termes entropiques qui reflètent le nombre de manières d’arranger toutes les entités présentes dans le milieu, qu’il s’agisse de segments de polymères ou de molécules de solvant. La contribution du polymère à l’entropie comporte un facteur en 1/N qui traduit la connectivité du polymère et donc le nombre moindre de possibilités d’arranger les segments polymères dans le réseau par comparaison au mélange de molécules simples. La plus grande difficulté à solubiliser les polymères par rapport à de petites molécules trouve là une première origine. La seconde cause de la solubilisation parfois difficile d’un polymère provient, quant à elle, de l’enthalpie de mélange (dernier terme de la formule) et, plus précisément, de l’énergie due aux contacts entre le polymère et le solvant. Cet aspect est pris en compte à travers le paramètre d’interaction, qui peut s’écrire:
où k est la constante de Boltzmann.Les deux premiers termes de cette formule sont des termes entropiques qui reflètent le nombre de manières d’arranger toutes les entités présentes dans le milieu, qu’il s’agisse de segments de polymères ou de molécules de solvant. La contribution du polymère à l’entropie comporte un facteur en 1/N qui traduit la connectivité du polymère et donc le nombre moindre de possibilités d’arranger les segments polymères dans le réseau par comparaison au mélange de molécules simples. La plus grande difficulté à solubiliser les polymères par rapport à de petites molécules trouve là une première origine. La seconde cause de la solubilisation parfois difficile d’un polymère provient, quant à elle, de l’enthalpie de mélange (dernier terme de la formule) et, plus précisément, de l’énergie due aux contacts entre le polymère et le solvant. Cet aspect est pris en compte à travers le paramètre d’interaction, qui peut s’écrire: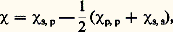 﨑s,p, 﨑p,p, 﨑s,s étant respectivement les énergies d’interaction solvant-polymère, polymère-polymère et solvant-solvant. 﨑 peut prendre des valeurs négatives si l’attraction polymère-solvant domine l’attraction solvant-solvant ou polymère-polymère et demeure positif dans le cas inverse; il est égal à 0,5 dans les conditions thêta. Dans la réalité, 﨑 est en général supérieur à 0. Le fait qu’il est parfois négatif traduit l’existence d’interactions spécifiques fortes, telles des liaisons hydrogène entre soluté et solvant.Telle qu’elle a été établie dans sa première version, la théorie de Flory-Huggins ne prévoit pas que le paramètre d’interaction 﨑 puisse croître ou décroître avec la température. Cela implique que le mélange polymère-solvant ne peut théoriquement se séparer en deux phases qu’à une température plus basse (upper critical solution temperature ) que celle de la miscibilité totale. Or il se trouve que les mélanges solvants-polymères peuvent aussi donner lieu à une séparation de phases lors d’un accroissement de température (lower critical solution temperature ). Une adaptation à la théorie de Flory-Huggins (dite théorie de Sanchez-Lacombe) faisant intervenir une possible compression du réseau par la présence de sites vacants (mailles libres), a permis de surmonter cette difficulté. La raison physique de l’immiscibilité à température élevée provient simplement des différences de coefficients d’expansion thermique des deux constituants du mélange qui, dès lors, deviennent incapables de constituer un mélange homogène en raison de densités trop contrastées.Ces quelques aménagements qui ont progressivement été apportés à la théorie de Flory-Huggins n’ôtent cependant rien à la pertinence de ce modèle, qui est aujourd’hui encore l’outil de référence pour la détermination de la masse molaire d’un échantillon à partir de sa solution. La mesure de la pression osmotique nous en donne une bonne illustration.Mesure de la pression osmotiqueLa pression osmotique 刺 exercée par les macromolécules en solution se définit comme la différence entre les potentiels chimiques du solvant dans la solution ( 猪1) et à l’état pur ( 猪01) divisée par le volume molaire du solvant (V1):
﨑s,p, 﨑p,p, 﨑s,s étant respectivement les énergies d’interaction solvant-polymère, polymère-polymère et solvant-solvant. 﨑 peut prendre des valeurs négatives si l’attraction polymère-solvant domine l’attraction solvant-solvant ou polymère-polymère et demeure positif dans le cas inverse; il est égal à 0,5 dans les conditions thêta. Dans la réalité, 﨑 est en général supérieur à 0. Le fait qu’il est parfois négatif traduit l’existence d’interactions spécifiques fortes, telles des liaisons hydrogène entre soluté et solvant.Telle qu’elle a été établie dans sa première version, la théorie de Flory-Huggins ne prévoit pas que le paramètre d’interaction 﨑 puisse croître ou décroître avec la température. Cela implique que le mélange polymère-solvant ne peut théoriquement se séparer en deux phases qu’à une température plus basse (upper critical solution temperature ) que celle de la miscibilité totale. Or il se trouve que les mélanges solvants-polymères peuvent aussi donner lieu à une séparation de phases lors d’un accroissement de température (lower critical solution temperature ). Une adaptation à la théorie de Flory-Huggins (dite théorie de Sanchez-Lacombe) faisant intervenir une possible compression du réseau par la présence de sites vacants (mailles libres), a permis de surmonter cette difficulté. La raison physique de l’immiscibilité à température élevée provient simplement des différences de coefficients d’expansion thermique des deux constituants du mélange qui, dès lors, deviennent incapables de constituer un mélange homogène en raison de densités trop contrastées.Ces quelques aménagements qui ont progressivement été apportés à la théorie de Flory-Huggins n’ôtent cependant rien à la pertinence de ce modèle, qui est aujourd’hui encore l’outil de référence pour la détermination de la masse molaire d’un échantillon à partir de sa solution. La mesure de la pression osmotique nous en donne une bonne illustration.Mesure de la pression osmotiqueLa pression osmotique 刺 exercée par les macromolécules en solution se définit comme la différence entre les potentiels chimiques du solvant dans la solution ( 猪1) et à l’état pur ( 猪01) divisée par le volume molaire du solvant (V1):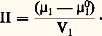 Cette pression osmotique est aussi reliée à l’energie libre par l’expression:
Cette pression osmotique est aussi reliée à l’energie libre par l’expression: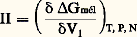 pour une température (T), une pression (P) et un nombre d’unités monomères (N) donnés.En faisant apparaître, dans l’expression de l’énergie libre, le coefficient du viriel A2 (définissant la qualité du solvant), puis en prenant sa dérivée, on aboutit à l’expression suivante de la pression osmotique:
pour une température (T), une pression (P) et un nombre d’unités monomères (N) donnés.En faisant apparaître, dans l’expression de l’énergie libre, le coefficient du viriel A2 (définissant la qualité du solvant), puis en prenant sa dérivée, on aboutit à l’expression suivante de la pression osmotique: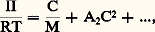 C étant la concentration du polymère, M sa masse molaire et R la constante des gaz parfaits. Comme la pression osmotique d’une solution est une propriété purement colligative, c’est-à-dire dépendant seulement du nombre de molécules et non de leur constitution chimique, sa mesure permet d’accéder à la masse molaire moyenne en nombre. A2 correspond à la pente de la droite qui caractérise la variation de 刺/C en fonction de C. Ce coefficient de viriel donne une idée de la qualité du solvant (si A2 礪 0, bon solvant; si A2 = 0, conditions thêta; si A2 麗 0, mauvais solvant). La variation de la pression osmotique avec la concentration (voir formule ci-dessus) n’est en réalité linéaire qu’en milieu dilué. En régime semi-dilué, 刺 ne varie pas de façon linéaire avec la concentration et est indépendante de la masse molaire du polymère. On retrouve là les arguments de De Gennes sur la limitation du phénomène de volume exclu aux distances 﨡 pour C 礪 C (effet d’écrantage). Les tenants de cette approche ont également proposé de prendre en compte le fait que les segments de la chaîne situés en dehors de 﨡 ne sont pas nécessairement non perturbés. Cela restreint les possibilités de placement des segments étrangers au voisinage du segment considéré, ce qui contribue à diminuer la pression osmotique à travers la concentration locale en segments. C’est ainsi qu’en milieu semi-dilué la formule de Des Cloizeaux prévoit que 刺 est proportionnelle à C9/4 et non à C2 comme nous venons de le voir. Les expériences montrent une corrélation excellente des résultats expérimentaux avec les prédictions du modèle de Des Cloizeaux. La théorie de Flory s’écarte d’autant plus de la réalité observée que la concentration du milieu s’accroît. La mesure de la pression osmotique est une technique puissante, car elle permet non seulement d’accéder à la masse molaire en nombre (face="EU Upmacr" 聾n) d’un échantillon (indépendamment de sa structure chimique), mais elle donne également des informations sur la qualité du solvant utilisé. Son peu de succès auprès de l’expérimentateur tient au fait qu’il est nécessaire de laisser suffisamment de temps à la solution pour qu’elle atteigne l’équilibre et trouver des membranes vraiment imperméables aux polymères et non sujettes à l’absorption. Cette technique est bien adaptée à la caractérisation des échantillons de masse molaire intermédiaire (104g/mol 麗 聾 麗 105g/mol).Diffusion de lumièreLa diffusion de lumière est aussi une technique précieuse pour l’expérimentateur, car elle livre de nombreux renseignements sur les solutions de polymères à caractériser. Outre la masse molaire d’un échantillon, il est possible de déterminer les dimensions et la forme des macromolécules et d’accéder au coefficient du viriel A2.Quand l’onde électromagnétique d’un faisceau monochromatique frappe une molécule, le dipôle, qui est induit par le champ E, vibre et constitue la source d’une radiation diffusante. Si la fréquence et la phase de la lumière diffusante sont les mêmes que celles de la lumière incidente, la diffusion est dite cohérente, ou élastique. L’expression du rapport de l’intensité diffusée à un angle [I( )] à l’intensité incidente (I0), connu sous le nom de rapport de Rayleigh (R size=1), s’écrit:
C étant la concentration du polymère, M sa masse molaire et R la constante des gaz parfaits. Comme la pression osmotique d’une solution est une propriété purement colligative, c’est-à-dire dépendant seulement du nombre de molécules et non de leur constitution chimique, sa mesure permet d’accéder à la masse molaire moyenne en nombre. A2 correspond à la pente de la droite qui caractérise la variation de 刺/C en fonction de C. Ce coefficient de viriel donne une idée de la qualité du solvant (si A2 礪 0, bon solvant; si A2 = 0, conditions thêta; si A2 麗 0, mauvais solvant). La variation de la pression osmotique avec la concentration (voir formule ci-dessus) n’est en réalité linéaire qu’en milieu dilué. En régime semi-dilué, 刺 ne varie pas de façon linéaire avec la concentration et est indépendante de la masse molaire du polymère. On retrouve là les arguments de De Gennes sur la limitation du phénomène de volume exclu aux distances 﨡 pour C 礪 C (effet d’écrantage). Les tenants de cette approche ont également proposé de prendre en compte le fait que les segments de la chaîne situés en dehors de 﨡 ne sont pas nécessairement non perturbés. Cela restreint les possibilités de placement des segments étrangers au voisinage du segment considéré, ce qui contribue à diminuer la pression osmotique à travers la concentration locale en segments. C’est ainsi qu’en milieu semi-dilué la formule de Des Cloizeaux prévoit que 刺 est proportionnelle à C9/4 et non à C2 comme nous venons de le voir. Les expériences montrent une corrélation excellente des résultats expérimentaux avec les prédictions du modèle de Des Cloizeaux. La théorie de Flory s’écarte d’autant plus de la réalité observée que la concentration du milieu s’accroît. La mesure de la pression osmotique est une technique puissante, car elle permet non seulement d’accéder à la masse molaire en nombre (face="EU Upmacr" 聾n) d’un échantillon (indépendamment de sa structure chimique), mais elle donne également des informations sur la qualité du solvant utilisé. Son peu de succès auprès de l’expérimentateur tient au fait qu’il est nécessaire de laisser suffisamment de temps à la solution pour qu’elle atteigne l’équilibre et trouver des membranes vraiment imperméables aux polymères et non sujettes à l’absorption. Cette technique est bien adaptée à la caractérisation des échantillons de masse molaire intermédiaire (104g/mol 麗 聾 麗 105g/mol).Diffusion de lumièreLa diffusion de lumière est aussi une technique précieuse pour l’expérimentateur, car elle livre de nombreux renseignements sur les solutions de polymères à caractériser. Outre la masse molaire d’un échantillon, il est possible de déterminer les dimensions et la forme des macromolécules et d’accéder au coefficient du viriel A2.Quand l’onde électromagnétique d’un faisceau monochromatique frappe une molécule, le dipôle, qui est induit par le champ E, vibre et constitue la source d’une radiation diffusante. Si la fréquence et la phase de la lumière diffusante sont les mêmes que celles de la lumière incidente, la diffusion est dite cohérente, ou élastique. L’expression du rapport de l’intensité diffusée à un angle [I( )] à l’intensité incidente (I0), connu sous le nom de rapport de Rayleigh (R size=1), s’écrit: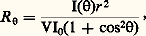 où r représente la distance entre le volume diffusant et le détecteur et V le volume diffusant.Ce rapport de Rayleigh est directement relié à la turbidité du système (concentration des polymères dans la solution); du point de vue des caractéristiques moléculaires de la solution, R size=1 peut être défini comme: R size=1 = KMC, où:
où r représente la distance entre le volume diffusant et le détecteur et V le volume diffusant.Ce rapport de Rayleigh est directement relié à la turbidité du système (concentration des polymères dans la solution); du point de vue des caractéristiques moléculaires de la solution, R size=1 peut être défini comme: R size=1 = KMC, où: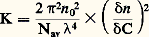 où n et n 0 représentent respectivement l’indice de réfraction du liquide et du solvant, la longueur d’onde de la lumière et av le nombre d’Avogadro. Cette équation montre que la masse molaire d’un échantillon peut être déterminée à dilution infinie:
où n et n 0 représentent respectivement l’indice de réfraction du liquide et du solvant, la longueur d’onde de la lumière et av le nombre d’Avogadro. Cette équation montre que la masse molaire d’un échantillon peut être déterminée à dilution infinie: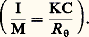 .Revenons à présent à l’origine du phénomène de diffusion par une solution. La diffusion provient des fluctuations locales de concentration qui engendrent des variations locales de polarisabilité et d’indice de réfraction. La théorie d’Einstein et de Smoluchowski permet de relier les fluctuations d’indice de réfraction à la variation de la pression osmotique:
.Revenons à présent à l’origine du phénomène de diffusion par une solution. La diffusion provient des fluctuations locales de concentration qui engendrent des variations locales de polarisabilité et d’indice de réfraction. La théorie d’Einstein et de Smoluchowski permet de relier les fluctuations d’indice de réfraction à la variation de la pression osmotique: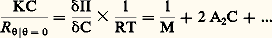 En raison de la relation entre la diffusion et la pression osmotique, il est possible, par diffusion de lumière, de mesurer la masse molaire d’un échantillon.Nous avons jusqu’à présent considéré que les particules étaient assimilables à des points et qu’il n’y avait donc pas de dépendance angulaire de l’intensité diffusée. Quand la taille des particules dépasse/20, l’intensité diffusée montre une certaine dépendance angulaire. La lumière diffusée est, en effet, affaiblie par l’interférence mutuelle entre les ondes diffusées par une même macromolécule. Un facteur P( ) est introduit pour tenir compte de cette dépendance angulaire :
En raison de la relation entre la diffusion et la pression osmotique, il est possible, par diffusion de lumière, de mesurer la masse molaire d’un échantillon.Nous avons jusqu’à présent considéré que les particules étaient assimilables à des points et qu’il n’y avait donc pas de dépendance angulaire de l’intensité diffusée. Quand la taille des particules dépasse/20, l’intensité diffusée montre une certaine dépendance angulaire. La lumière diffusée est, en effet, affaiblie par l’interférence mutuelle entre les ondes diffusées par une même macromolécule. Un facteur P( ) est introduit pour tenir compte de cette dépendance angulaire :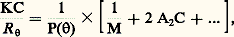 avec:
avec: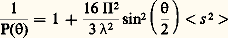 麗礪ce qui donne, dans la limite de C0:
麗礪ce qui donne, dans la limite de C0: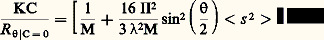 Cette expression montre que la masse molaire (dont Flory a montré qu’il s’agissait de 聾w) peut être calculée par double extrapolation à C et = 0. Connaissant alors la valeur de M, on peut en déduire le carré moyen du rayon de giration 麗s2 礪 résultant, lui, de l’extrapolation à C = 0.Viscosité intrinsèqueSi la pression osmotique et la diffusion de lumière sont directement conditionnées pour la première par le nombre de molécules présentes et la deuxième par la masse molaire, la viscosité intrinsèque et la chromatographie par perméation de gel (GPC) sont, elles, sensibles au rayon de giration de la macromolécule. Ces deux techniques présentent l’avantage d’être faciles à mettre en œuvre, mais elles ne constituent en aucune manière des méthodes absolues de détermination des masses molaires. L’analyse de la viscosité d’une solution de polymère est fondée sur l’observation que la viscosité d’un liquide dans lequel une particule est introduite s’accroît proportionnellement au volume de cette particule. C’est ainsi que la théorie d’Einstein prévoit que la viscosité d’une suspension diluée de sphères dures s’écrit:
Cette expression montre que la masse molaire (dont Flory a montré qu’il s’agissait de 聾w) peut être calculée par double extrapolation à C et = 0. Connaissant alors la valeur de M, on peut en déduire le carré moyen du rayon de giration 麗s2 礪 résultant, lui, de l’extrapolation à C = 0.Viscosité intrinsèqueSi la pression osmotique et la diffusion de lumière sont directement conditionnées pour la première par le nombre de molécules présentes et la deuxième par la masse molaire, la viscosité intrinsèque et la chromatographie par perméation de gel (GPC) sont, elles, sensibles au rayon de giration de la macromolécule. Ces deux techniques présentent l’avantage d’être faciles à mettre en œuvre, mais elles ne constituent en aucune manière des méthodes absolues de détermination des masses molaires. L’analyse de la viscosité d’une solution de polymère est fondée sur l’observation que la viscosité d’un liquide dans lequel une particule est introduite s’accroît proportionnellement au volume de cette particule. C’est ainsi que la théorie d’Einstein prévoit que la viscosité d’une suspension diluée de sphères dures s’écrit: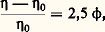 兀0 étant la viscosité du solvant, 兀 celle de la solution et 﨏 la fraction volumique de ces sphères.La fraction volumique peut s’écrire:
兀0 étant la viscosité du solvant, 兀 celle de la solution et 﨏 la fraction volumique de ces sphères.La fraction volumique peut s’écrire: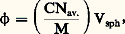 où C est la concentration du polymère et Vsph le volume effectif de la chaîne. Comme le volume et le rayon de giration sont reliés sous la forme Vsph = k’ s3, la viscosité intrinsèque [ 兀] peut s’écrire:
où C est la concentration du polymère et Vsph le volume effectif de la chaîne. Comme le volume et le rayon de giration sont reliés sous la forme Vsph = k’ s3, la viscosité intrinsèque [ 兀] peut s’écrire: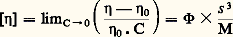 avec 﨏 = 2,5 k ’av. (k’ étant une constante).Cette équation, dite de Flory-Fox, montre clairement la relation qui existe entre la viscosité intrinsèque [ 兀] et le rayon de giration. Mesurer la viscosité intrinsèque donne ainsi accès au volume occupé par la molécule dans l’espace. 﨏 est, quant à elle, une constante dont la valeur est approximativement égale à 2,25 憐 1023 par mole.Par ailleurs, la viscosité intrinsèque et la masse molaire d’un échantillon ont été reliées de façon empirique sous la forme d’une équation dite de Mark-Houwink-Sakurada: [ 兀] = KMa , où K est une constante et a le coefficient de Mark-Houwink-Sakurada qui varie entre 0,5 (conditions thêta) et 0,8 (bon solvant).De façon pratique, la viscosité intrinsèque est obtenue en extrapolant à concentration nulle la variation de:
avec 﨏 = 2,5 k ’av. (k’ étant une constante).Cette équation, dite de Flory-Fox, montre clairement la relation qui existe entre la viscosité intrinsèque [ 兀] et le rayon de giration. Mesurer la viscosité intrinsèque donne ainsi accès au volume occupé par la molécule dans l’espace. 﨏 est, quant à elle, une constante dont la valeur est approximativement égale à 2,25 憐 1023 par mole.Par ailleurs, la viscosité intrinsèque et la masse molaire d’un échantillon ont été reliées de façon empirique sous la forme d’une équation dite de Mark-Houwink-Sakurada: [ 兀] = KMa , où K est une constante et a le coefficient de Mark-Houwink-Sakurada qui varie entre 0,5 (conditions thêta) et 0,8 (bon solvant).De façon pratique, la viscosité intrinsèque est obtenue en extrapolant à concentration nulle la variation de: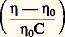 en fonction de C.Chromatographie par perméation de gelLa chromatographie par perméation de gel (GPC) est certainement la méthode la plus employée et la plus appréciée de l’expérimentateur. À l’instar de la viscosité, la GPC est avant tout sensible au rayon de giration de la molécule. Le principe de la GPC est fondé sur la séparation de macromolécules par leur élution (transport d’un solvant d’une macromolécule à travers le milieu) à travers une colonne remplie de gel poreux ou de silice microporeuse. L’échantillon, qui est injecté dans cette colonne puis élué sous un flux de solvant, y séjourne pendant un laps de temps qui dépend du volume des pores qui lui ont été accessibles dans le gel. Le solvant a, quant à lui, accès à tous les pores du gel. Pour qu’une colonne puisse séparer une population de macromolécules de façon efficace, la taille des pores les plus grands doit être distribuée de façon large et leur volume légèrement supérieur à celui de la fraction de polymère qui présente la masse molaire la plus élevée. Le volume d’élution Ve peut s’écrire:
en fonction de C.Chromatographie par perméation de gelLa chromatographie par perméation de gel (GPC) est certainement la méthode la plus employée et la plus appréciée de l’expérimentateur. À l’instar de la viscosité, la GPC est avant tout sensible au rayon de giration de la molécule. Le principe de la GPC est fondé sur la séparation de macromolécules par leur élution (transport d’un solvant d’une macromolécule à travers le milieu) à travers une colonne remplie de gel poreux ou de silice microporeuse. L’échantillon, qui est injecté dans cette colonne puis élué sous un flux de solvant, y séjourne pendant un laps de temps qui dépend du volume des pores qui lui ont été accessibles dans le gel. Le solvant a, quant à lui, accès à tous les pores du gel. Pour qu’une colonne puisse séparer une population de macromolécules de façon efficace, la taille des pores les plus grands doit être distribuée de façon large et leur volume légèrement supérieur à celui de la fraction de polymère qui présente la masse molaire la plus élevée. Le volume d’élution Ve peut s’écrire: où Vi est le volume intersticiel du gel, Vp le volume des pores du gel et K le coefficient de partition de la porosité de la colonne.Le fait que K soit conditionné par le volume des pores de la colonne et par le volume hydrodynamique des chaînes (s3 = [ 兀] 憐 M), a été clairement mis en évidence par H. Benoit et ses collaborateurs. L’excellente corrélation entre [ 兀] 憐 M et le temps d’élution (indépendamment du type d’échantillon considéré) a ainsi été montrée par ces auteurs à travers l’établissement d’une courbe d’étalonnage universel. Même si la GPC n’est pas une méthode absolue de mesure des masses molaires et même si elle requiert un étalonnage préalable par des échantillons de masse molaire connue, elle est précieuse, car elle donne accès à une information capitale pour l’expérimentateur, à savoir la distribution des masses molaires dans un échantillon.4. Viscoélasticité des macromoléculesRhéologie des polymèresÀ l’état fondu ou à concentration élevée, le comportement des macromolécules est conditionné, plus que pour tout autre état, par leur masse molaire. Les pelotes macromoléculaires de dimension moléculaires suffisantes peuvent s’enchevêtrer et, dès lors, totalement changer les propriétés d’écoulement. À cet égard, l’étude du flux et des propriétés viscoélastiques des polymères en solution ou à l’état fondu apporte de précieuses informations sur la structure des polymères. La discipline qui traite des relations entre la contrainte et la déformation d’un liquide est appelée rhéologie. Un liquide polymère (en particulier à l’état fondu) est caractérisé par un comportement viscoélastique; l’analyse rhéologique a, en réalité, trait à l’étude du comportement de ce liquide polymère au-delà des limites d’une vitesse de cisaillement nulle et d’une dilution infinie, qui concernent, quant à elles, plus particulièrement la viscosité intrinsèque. Les mesures rhéologiques peuvent être effectuées de diverses manières, notamment par la détermination de la contrainte subie par le milieu en réponse à une déformation constante ou, au contraire, par la sollicitation de façon constante du milieu et examen de la déformation engendrée.Trois grandeurs peuvent être obtenues au terme d’une expérience de rhéologie:– la viscosité ( 兀), qui est le rapport de la contrainte de cisaillement à la vitesse de cisaillement;– le module de conservation (G ), qui est le rapport de la composante en phase de la contrainte sur la déformation sinusoïdale;– le module de perte (G ), qui est le rapport de la composante déphasée de 900 de la contrainte sur cette même déformation sinusoïdale.G caractérise l’élasticité du matériau alors que G traduit la dissipation de l’énergie par friction.Dans leur très grande majorité, les polymères ne se comportent pas comme des liquides newtoniens, lesquels sont caractérisés par une viscosité constante indépendante de la vitesse de cisaillement appliquée. Un liquide polymère manifeste, en réalité, toutes les caractéristiques d’un liquide pseudoplastique, c’est-à-dire qu’après un premier plateau newtonien sa viscosité aura tendance à baisser à mesure que la vitesse de cisaillement augmente. Aux vitesses de cisaillement les plus faibles, les chaînes présentent des conformations peu différentes de celle de l’état non perturbé et il est probable que les enchevêtrements contribuent pleinement à la résistance à l’écoulement. Avec l’augmentation de la vitesse de cisaillement, les macromolécules s’alignent dans la direction du cisaillement, prennent une forme anisotrope et se contractent dans la direction perpendiculaire à l’axe du cisaillement. Une autre caractéristique des liquides polymères est leur propension à se comporter comme des liquides thixotropes: à vitesse de cisaillement constante, leur viscosité diminue en effet avec le temps d’application de la contrainte.À l’instar du temps d’application de la contrainte ou de la vitesse de cisaillement, la concentration du polymère dans le milieu peut aussi grandement affecter la viscosité.Au-delà d’une certaine concentration d’espèce de masse molaire (Mc), appelée masse molaire critique, la viscosité peut augmenter de façon abrupte, traduisant la présence d’interactions intermoléculaires (enchevêtrements). Même s’il n’existe aucune théorie capable de prévoir l’apparition du phénomène d’enchevêtrement, on peut en donner une explication empirique. Tant que la masse molaire d’un échantillon est inférieure à Mc, la viscosité est essentiellement gouvernée par le coefficient de friction (f ). La viscosité est dans ce cas proportionnelle à C[ 兀]. Au-delà de Mc et en présence d’enchevêtrements, la coopérativité que requièrent les longues chaînes pour se mouvoir l’une par rapport à l’autre freine leur diffusion; la viscosité varie alors en C 憐 聾w.L’existence d’enchevêtrements peut ainsi être décelée à travers la mesure de G’ qui, dans son augmentation avec la fréquence de la déformation sinuso 練dale, marque un plateau avant de croître à nouveau. La masse molaire (Me) à partir de laquelle ces enchevêtrements se manifestent est empiriquement 1/2 Mc. On dit que les enchevêtrements sont des points de réticulation temporaire.Interprétation théorique du comportement rhéologiqueLe premier modèle proposé est celui de Rouse (1953). Il représente la chaîne comme un assemblage de perles reliées entre elles par des ressorts. Ces perles sont censées interagir par friction (avec un coefficient f ) avec leur milieu environnant, tandis que les ressorts répondent de façon élastique à une sollicitation. Le milieu visqueux exerçant une force d’entraînement sur la chaîne (force proportionnelle à la vitesse des perles), la solution des équations de vitesse des perles donne accès aux temps de relaxation caractéristiques des modes internes de mouvement de la chaîne. En réalité ni ce modèle ni sa version améliorée, qui tient compte de la force exercée par les unités répétitives adjacentes ainsi que du mouvement brownien de la chaîne, ne prennent en compte les enchevêtrements. Ces modèles, qui ignorent également la variation de la viscosité avec la vitesse de cisaillement, sont en fait bien appropriés pour les milieux dilués.La description qui répond le mieux aux réalités auxquelles est confronté l’expérimentateur est celle proposée par de Gennes à travers son concept de reptation [cf. GENNES (P.-G. de)]. Selon ce modèle, la chaîne confrontée aux obstacles que constituent les enchevêtrements se meut à l’intérieur d’un tube qui représente approximativement son contour, comme le ferait un serpent à travers les obstacles sans les franchir vraiment. À côté de ce mouvement longitudinal de désengagement du tube, le modèle envisage également la possibilité de changements conformationnels à l’intérieur même du tube. Cette théorie prédit que la viscosité est très dépendante de la concentration du milieu et de la masse molaire de l’échantillon ( 兀 théorique est proportionnelle à M3 alors que 兀 mesurée est proportionnelle à M3,4). Même si résultats expérimentaux et prédictions théoriques ne concordent pas toujours très bien, la meilleure preuve de la réalité du phénomène de reptation est à trouver dans la différence entre les polymères linéaires et les polymères ramifiés, ces derniers diffusant effectivement beaucoup plus lentement.Quatre régions de viscoélasticité en fonction de la températureSi l’étude du comportement des solutions de fluides polymères est déterminante pour la connaissance des caractéristiques moléculaires d’un échantillon, pour sa mise en forme, pour sa rhéologie ou encore pour la réalisation de quelques applications bien ciblées, il est aussi essentiel de cerner les propriétés des polymères à l’état solide pour pouvoir les utiliser comme matériaux.À une température suffisamment basse, tout échantillon polymère est rigide, dur et se déforme de façon essentiellement élastique (dans la limite des faibles déformations), indépendamment du type d’organisation qu’adoptent ses chaînes: organisation totalement amorphe ou semi-cristalline. Le fait que certaines chaînes ne sont pas en mesure de cristalliser est essentiellement dû aux irrégularités et aux imperfections de leur structure. À mesure que la température s’accroît, le polymère a tendance à se comporter comme un matériau viscoélastique, réunissant les caractéristiques d’un liquide visqueux et celles d’un solide élastique. C’est à travers l’évolution des propriétés mécaniques que les phénomènes de viscoélasticité peuvent être le mieux mises en évidence. Dans un tel matériau viscoélastique, les propriétés mécaniques, et en particulier le module d’élasticité, dépendent de la durée d’application de la contrainte et de la température à laquelle s’applique la contrainte. Deux phénomènes caractéristiques se manifestent:– la relaxation, qui correspond à la diminution de la contrainte nécessaire pour maintenir une déformation constante au cours du temps;– le fluage, qui traduit l’augmentation de la déformation à contrainte constante.La relaxation se réfère, au plan moléculaire, au temps que mettent les macromolécules à répondre à une sollicitation extérieure par un mouvement d’orientation dans la direction de la contrainte. Quant au fluage, il résulte du glissement des chaînes les unes par rapport aux autres à tension constante.Quatre régions principales peuvent être distinguées au cours de l’étude viscoélastique d’un matériau polymère:– La première région correspond à l’état vitreux (état solide). Le module d’élasticité est de l’ordre de 3 憐 109 Pa indépendamment de la nature chimique du polymère. Les mouvements portent sur des distances très courtes et se limitent à la vibration, voire à la rotation d’éléments locaux de la chaîne. Des relaxations secondaires, concernant en particulier des groupes latéraux à la chaîne principale, ont pu être mises en évidence sur des polymères tels que le poly(méthacrylate de méthyle).– La deuxième région est celle de la transition vitreuse. Plusieurs théories ont été proposées pour rendre compte du phénomène de transition vitreuse. La première est celle dite du volume libre, selon laquelle le volume de matière inoccupée (ou libre) est invariable au-dessous de la température de transition vitreuse (Tg). Un deuxième groupe de théories décrit la transition vitreuse comme un phénomène purement cinétique qui se manifeste dès lors que le temps de réponse à une sollicitation est du même ordre de grandeur que le temps caractéristique de l’expérience. Enfin la théorie de Gibbs-Di Marzio fonde l’existence de la transition vitreuse sur le postulat que l’entropie de conformation est égale à O à Tg, même si les auteurs reconnaissent l’aspect cinétique du phénomène. Cependant, aucune de ces théories n’est vraiment en mesure d’expliquer de façon satisfaisante la transition vitreuse. Quoi qu’il en soit, la température à laquelle intervient cette transition dépend de la nature chimique de la chaîne macromoléculaire. Le module de conservation G’ décroît d’un facteur 103 pour une variation de 20 à 30 0C: la région de la transition vitreuse peut être qualitativement définie comme celle où apparaissent des mouvements coordonnés sur de longues distances.– La troisième région de viscoélasticité est celle du plateau caoutchoutique. Le polymère présente le comportement typique d’un caoutchouc avec des mouvements coordonnés sur de longues distances. Le module d’élasticité prend des valeurs de l’ordre de 2 憐 106 Pa et le plateau caoutchoutique est d’autant plus étendu que les chaînes sont longues. Si l’échantillon est réticulé, ce plateau caoutchoutique peut s’étendre sur une grande plage de température.– La quatrième et dernière région est celle de l’écoulement. À des températures plus élevées encore, l’énergie fournie permet aux chaînes de serpenter à travers les enchevêtrements et de s’écouler.Le comportement observé pour les échantillons semi-cristallins diffère quelque peu de celui des matériaux amorphes. Comme la température de fusion des parties cristallines est toujours supérieure à celle de la transition vitreuse dans les polymères semi-cristallins, le module d’élasticité prend des valeurs qui dépendent du degré de cristallinité des chaînes. Au-delà de la température de fusion des zones cristallines, le polymère s’écoule à l’instar d’un matériau amorphe.Cette partie concernant les propriétés viscoélastiques des matériaux polymères à l’état solide ne saurait être complète sans que soit évoquée le principe d’équivalence temps-température. Selon ce principe, le temps et la température sont deux grandeurs équivalentes pour les matériaux viscoélastiques. A partir de portions de courbe donnant l’évolution du module de conservation G’ en fonction du temps et à différentes températures, il est ainsi possible de tracer une courbe dite maîtresse à une température de référence par translation et superposition de ces différentes parties de courbe. Cette courbe maîtresse serait impossible à établir pour la majorité des polymères, par exemple à température ambiante, pour des raisons techniques: la plage de fréquences de sollicitation disponibles sur les appareillages est en effet restreinte. Le comportement d’un matériau viscoélastique sollicité à température élevée pendant des temps courts est ainsi comparable à celui du même matériau à basse température et pendant des temps plus longs. En d’autres termes, un changement de température produit le même effet que l’application d’un facteur multiplicatif à l’échelle des temps.Bien que jeune, la science des polymères a connu un développement constant depuis les travaux de H. Standinger dans les années 1920, qui s’est concrétisé par une multiplicité d’applications dans des domaines très divers. De nouvelles avancées ne pourraient résulter que d’une fructueuse collaboration entre chimistes et physiciens.5. Les macromolécules biologiquesTous les êtres vivants sont constitués de cellules, d’une seule, comme chez les bactéries ou les algues primitives; ou de plusieurs, environ 1015, comme chez les mammifères. Il est généralement admis que tous les êtres vivants dérivent d’une seule cellule primitive, apparue sur la Terre il y a environ 3,5 milliards d’années. Cette cellule s’est reproduite et les descendants ont évolué. Aujourd’hui, il existe deux grands types de cellules, la cellule eucaryote, qui possède un noyau, et la cellule procaryote, qui n’en possède pas. Ces cellules présentent beaucoup d’autres différences, comme la taille, l’organisation, la complexité, etc., mais se ressemblent par la nature des substances chimiques qu’elles contiennent et par leurs quantités relatives (tabl. 1). Le constituant principal des cellules est l’eau. En plus des ions inorganiques, les autres constituants, tous formés d’un squelette carboné [cf. ORGANIQUE (CHIMIE)], peuvent être divisés en deux grands groupes en fonction de leur masse molaire. Le premier groupe comprend des molécules de faible masse molaire inférieure à 2 000 grammes par mole (limite arbitraire). Ce sont essentiellement les sucres, les acides gras, les acides aminés, les nucléotides et tous les précurseurs et intermédiaires du métabolisme – au total, près de 750 espèces différentes. Le second groupe comprend les molécules de grande masse molaire appelées macromolécules ou biopolymères. Les biopolymères (de l’ordre de 2 000 espèces différentes dans une bactérie) sont les protéines, les acides nucléiques et les polysaccharides, formés par l’union de motifs élémentaires ou monomères que sont les acides aminés, les nucléotides et les sucres, respectivement. Les lipides et les phospholipides ne sont pas des macromolécules, bien qu’ils puissent s’associer et former de très grosses particules. Dans une macromolécule, les monomères (identiques ou différents) sont liés entre eux par des liaisons covalentes (interactions fortes), alors que l’association des phospholipides et des lipides résultent d’interactions beaucoup plus faibles (d’origines électrostatique et hydrophobe).Les biopolymères ont un rôle essentiel dans le fonctionnement des cellules. Les progrès considérables réalisés dans tous les domaines scientifiques et techniques ont permis d’isoler un grand nombre de macromolécules biologiques et de déterminer, de façon très précise, leurs masses molaires, leurs formes globales et locales (dans certains cas à l’échelle atomique) et leurs fonctions biologiques.Les acides nucléiquesDeux familles d’acides nucléiques, l’acide désoxyribonucléique (ADN) et l’acide ribonucléique (ARN), sont présentes dans les cellules. L’ADN et l’ARN résultent de l’enchaînement linéaire régulier de nucléotides (séquences). Chaque nucléotide est constitué d’une base hétérocyclique azotée (adénine, guanine, cytosine ou thymine dans le cas de l’ADN; adénine, guanine, cytosine et uracile dans le cas de l’ARN), d’un sucre et d’un groupement phosphate. du point de vue chimique, la différence principale, apparemment minime, entre les nucléotides de l’ADN et l’ARN porte sur la nature du sucre, un désoxyribose et un ribose, respectivement. Néanmoins, l’ADN et l’ARN ont des structures et des fonctions tout à fait différentes.Dans la cellule, l’ADN adopte une structure en double hélice droite, résultant de l’association non covalente de deux brins (ou chaînes) complémentaires; chaque nucléotide d’un brin étant apparié à un nucléotide de l’autre brin. Cette structure a été décrite pour la première fois, en 1953, par F. H. C. Crick et par J. D. Watson. La masse molaire de l’ADN, très grande, varie suivant les espèces. L’ADN humain contient 3 milliards de paires de nucléotides. Si cet ADN pouvait être complètement étiré, il aurait une longueur d’environ 2 mètres pour un diamètre de l’ordre de 2 憐 10—9 mètres. De nombreuses études sur des fragments d’ADN, naturels ou synthétiques, ont démontré le polymorphisme de l’ADN. Suivant les conditions expérimentales, un fragment d’ADN adopte une structure en double hélice droite (avec des variantes dans les paramètres hélicoïdaux) ou une structure en double hélice gauche ou même des structures hélicoïdales plus complexes contenant trois ou quatre brins. Aussi bien sur une paire de nucléotides que sur plusieurs centaines ou milliers de nucléotides, la structure de l’ADN est dynamique et non statique.L’ADN est le support de l’information génétique (dans certains cas comme les rétrovirus, l’information génétique est portée par l’ARN, mais l’insertion de cette information dans l’ADN de la cellule hôte ne se fera qu’après copie de l’ARN en ADN par une enzye, la transcriptase inverse). Les ordres qui déterminent la nature et le nombre de la quasi-totalité des macromolécules intervenant dans le métabolisme cellulaire proviennent de l’ADN et sont transmis par l’intermédiaire d’une autre macromolécule, l’ARN messager. L’information génétique est conservée intégralement au cours de la division de la cellule mère en deux cellules filles. La cellule est capable de synthétiser deux copies conformes de son ADN (si l’on excepte les mutations), grâce à la complémentarité des deux brins de la double hélice [cf. NUCLÉIQUES (ACIDES)]. Cependant, il serait erroné de penser que le rôle de l’ADN n’est qu’informatif. L’ADN matrice interfère, par sa structure dynamique, avec l’action de protéines comme celles qui sont impliquées dans la réplication (synthèse d’ADN) et dans la transcription (synthèse de l’ARN).La transcription de l’ADN en ARN donne naissance à trois grandes classes d’ARN qui englobent les ARN messagers, les ARN ribosomaux et les ARN de transfert (chez les eucaryotes, d’autres ARN interviennent dans le métabolisme nucléaire).Comme l’ADN, les ARN ont une structure bien définie mais, contrairement à l’ADN, cette structure ne met pas en jeu l’association de plusieurs chaînes. Chaque chaîne d’ARN se replie sur elle-même avec appariement de nucléotides complémentaires situés à plus ou moins grande distance le long de la chaîne (modèle multi-épingle à cheveux ou en multi-tige boucle). Le repliement tridimensionnel de la chaîne dépend de la séquence nucléotidique et conduit donc à une structure propre à chaque chaîne d’ARN.Les trois classes d’ARN ont des fonctions différentes mais leur couplage dirige la synthèse protéique. La synthèse d’une protéine nécessite l’assemblage entre un ARN messager (qui porte l’instruction définissant la séquence de la protéine qu’il code), les ribosomes (édifices formés par l’association des ARN ribosomaux et de protéines permettant l’enchaînement des acides aminés) et les ARN de transfert (chaque sorte d’acide aminé étant fixé à un ARN de transfert spécifique).Pendant longtemps, il a été accepté que seules les protéines possédaient des propriétés catalytiques. Dans les années 1980, on a montré que des séquences d’ARN sont capables de catalyser la coupure et la formation de liaisons covalentes entre nucléotides. Leur existence permet d’expliquer plusieurs processus biologiques, et ouvrent ainsi de nouvelles perspectives dans la compréhension des phénomènes prébiotiques, c’est-à-dire ceux qui ont permis l’origine de la vie [cf. ORIGINE DE LA VIE]: l’ARN serait apparu avant l’ADN et aurait joué à la fois les deux rôles de porteur de l’information génétique et d’enzyme. Ces ARN catalytiques, appelés ribozomes, sont aussi efficaces dans le tube à essai et en absence de protéines.Les protéinesLes protéines constituent une classe de macromolécules d’une grande complexité. Elles résultent de l’enchaînement linéaire régulier d’acides aminés. Vingt sortes d’acides aminés, différant par la nature chimique de leurs chaînes latérales, entrent dans la composition des protéines, d’où une très grande diversité des protéines (l’enchaînement de 100 acides aminés différents permet de générer 20100 protéines). Les masses molaires des protéines couvrent un très large domaine, mais en général sont inférieures à 100 000 grammes par mole. En se basant sur la forme globale, on peut diviser les protéines en deux grands groupes: les protéines globulaires (sphères ou ellipsoïdes) et les protéines fibreuses (bâtonnets). Dans tous les cas, la composition en acides aminés et la séquence de la protéine déterminent la structure et la fonction de la protéine. Comprendre comment une protéine adopte une structure tridimensionnelle fonctionnelle fait encore à l’heure actuelle l’objet de très nombreux travaux.Le repliement d’une protéine résulte d’interactions faibles (non covalentes) entre les acides aminés. Ces interactions faibles stabilisent localement des structures ordonnées (hélice 見 et feuillet 廓) qui contiennent un nombre variable d’acides aminés. Les hélices 見 et les feuillets 廓 interagissent entre eux pour former des unités globulaires (domaines) qui à leur tour peuvent interagir entre elles. Enfin, plusieurs protéines (deux ou plus, identiques ou pas) peuvent s’associer pour former des édifices complexes actifs de grandes masses molaires. Il est remarquable que la structure tridimensionnelle des protéines, stabilisée par des interactions faibles, soit aussi précise. Le remplacement d’un acide aminé par un autre (mutagenèse) peut entraîner la modification de la structure de la protéine. Souvent, les protéines subissent des modifications post-traductionnelles spécifiques, comme la phosphorylation ou la fixation covalente de groupes carbohydrates (ces protéines sont alors appelées des glycoprotéines) ou d’acides gras (lipoprotéines).Les protéines sont les agents actifs de la cellule. Elles interviennent à tous les niveaux (rigidité structurale, perméabilité des membranes, transport, concentration des métabolites, déplacement, fonctionnement des gènes, etc.). Certaines d’entre elles, appelées enzymes, ont la propriété remarquable d’être des catalyseurs spécifiques. Un catalyseur augmente la vitesse d’une réaction sans changer ni la direction de la réaction ni le rapport à l’équilibre entre les produits et les réactifs et se retrouve intact à la fin de la réaction. Les enzymes sont capables d’augmenter la vitesse d’une réaction de 106 à 1012 fois par rapport à celle de la même réaction non catalysée et dans les mêmes conditions expérimentales. De plus, elles sont très spécifiques, en ce sens qu’elles sont capables de différencier des substrats de formules chimiques voisines. Enfin, les réactions sont souvent coordonnées sous l’action de complexes multienzymatiques. Les enzymes permettent de réaliser, dans une bactérie, plus de 3 000 réactions chimiques différentes, intégrées dans un ordre spatial et temporel précis et controlé.PolysaccharidesLes polysaccharides résultent de l’enchaînement linéaire ou ramifié de sucres. Bien qu’une quinzaine de sortes de sucres entrent dans la composition des polysaccharides, chaque polysaccharide ne contient qu’un nombre limité (de 1 à 6) de sortes de sucres. Tout en ayant la même composition chimique, des polysaccharides peuvent être différents. C’est le cas de l’amylose, de l’amylopectine et du glycogène, qui sont des polymères de l’ 見-D-glucopyranose. L’un de ces polymères – qui diffèrent par la position des liaisons covalentes entre les monomères – est linéaire (amylose), les deux autres sont ramifiés (amylopectine et glycogène). Contrairement aux acides nucléiques et aux protéines, qui ont presque toujours une masse molaire définie, les masses molaires des polysaccharides sont souvent variables. Les polysaccharides peuvent se structurer mais pas de façon aussi remarquable et variée que les protéines. En plus de leur position, l’orientation des liaisons covalentes entre monomères dans des homopolymères conduit à des structures différentes. L’amylose a une structure lâche alors que la cellulose a une structure très ordonnée. Toutes ces possibilités dans l’enchaînement des monomères rendent très compliquée la caractérisation physico-chimique des polysaccharides.Certains polysaccharides servent de stocks de matières nutritives. C’est le cas du glycogène dans les cellules animales et de l’amidon dans les cellules végétales: ces réserves de glucides sont décomposables lors du métabolisme (glycolyse). D’autres polysaccharides interviennent dans la construction des parois des cellules végétales et bactériennes. Enfin, de courts polysaccharides (oligosaccharides) greffés sur certaines macromolécules jouent un rôle très important dans les phénomènes de signalisation qui entrent en jeu au cours de l’activité cellulaire.Remarques sur les macromolécules biologiquesDe nombreuses études sont encore consacrées à l’élucidation de la structure et de la fonction de biopolymères dans des milieux physiquement simples (solutions diluées, cristaux...). L’intérêt de ces études s’est trouvé conforté parce que, en parallèle, il est vérifié que des réactions se produisant dans la cellule peuvent être reproduites, au moins partiellement, dans le tube à essai en utilisant un mélange adéquat de biopolymères purifiés et de petites molécules. Il est ainsi possible de répliquer et de transcrire l’ADN, de synthétiser des protéines, toutes ces réactions obéissant aux lois classiques de la physique et de la chimie. Cependant, les équilibres réactionnels ainsi que les vitesses des réactions peuvent être complètement modifiés dans la cellule. En effet, le milieu cellulaire est hautement organisé; les biopolymères sont confinés dans un espace restreint (d’où une concentration élevée) et les réactions sont couplées. Dans des solutions très concentrées de complexes ADN-protéines (conditions se rapprochant de celles qui sont rencontrées dans le noyau des cellules eucaryotes, bien que les concentrations ioniques, le pH et le degré d’hydratation soient peu connus), d’une part les propriétés fonctionnelles des complexes sont stimulées et, d’autre part, les interactions entre ces complexes donnent naissance à une organisation cristalline liquide. Il est certain que les problèmes d’organisation à l’échelle supramoléculaire dans les êtres vivants interviennent à plusieurs niveaux (membranes, cellule elle-même, tissus cellulaires, etc.) et jouent un rôle fondamental dans le fonctionnement biochimique.La complexité des êtres vivants est telle que, pour l’aborder, les scientifiques de différentes disciplines doivent mettre leurs compétences en commun. Réciproquement, la meilleure connaissance des propriétés des biopolymères et de leurs fonctions dans la cellule est à la base de la conception et de la synthèse de systèmes de plus en plus complexes, présentant des propriétés ressemblant de mieux en mieux à celles du vivant.Les études physico-chimiques des macromolécules biologiques ont joué un rôle prépondérant dans l’impressionnant développement des sciences biologiques durant la seconde moitié du XXe siècle. Ce développement entraîne des bouleversements profonds pour l’homme et son environnement. Chaque découverte ouvre de nouvelles possibilités de la maîtrise du vivant. En 1996, pour la première fois dans l’histoire des sciences, un mammifère a été créé à partir du noyau d’une cellule somatique prélevée sur un animal adulte. Être capable de cloner un mammifère adulte a une portée considérable, tant du point de vue fondamental qu’appliqué, mais ouvre aussi la perspective de l’utilisation de ces techniques de reproduction à l’homme. Devant un problème d’une telle ampleur, une réflexion entre toutes les communautés est absolument nécessaire pour définir les droits et les devoirs de l’homme face à la science.
où Vi est le volume intersticiel du gel, Vp le volume des pores du gel et K le coefficient de partition de la porosité de la colonne.Le fait que K soit conditionné par le volume des pores de la colonne et par le volume hydrodynamique des chaînes (s3 = [ 兀] 憐 M), a été clairement mis en évidence par H. Benoit et ses collaborateurs. L’excellente corrélation entre [ 兀] 憐 M et le temps d’élution (indépendamment du type d’échantillon considéré) a ainsi été montrée par ces auteurs à travers l’établissement d’une courbe d’étalonnage universel. Même si la GPC n’est pas une méthode absolue de mesure des masses molaires et même si elle requiert un étalonnage préalable par des échantillons de masse molaire connue, elle est précieuse, car elle donne accès à une information capitale pour l’expérimentateur, à savoir la distribution des masses molaires dans un échantillon.4. Viscoélasticité des macromoléculesRhéologie des polymèresÀ l’état fondu ou à concentration élevée, le comportement des macromolécules est conditionné, plus que pour tout autre état, par leur masse molaire. Les pelotes macromoléculaires de dimension moléculaires suffisantes peuvent s’enchevêtrer et, dès lors, totalement changer les propriétés d’écoulement. À cet égard, l’étude du flux et des propriétés viscoélastiques des polymères en solution ou à l’état fondu apporte de précieuses informations sur la structure des polymères. La discipline qui traite des relations entre la contrainte et la déformation d’un liquide est appelée rhéologie. Un liquide polymère (en particulier à l’état fondu) est caractérisé par un comportement viscoélastique; l’analyse rhéologique a, en réalité, trait à l’étude du comportement de ce liquide polymère au-delà des limites d’une vitesse de cisaillement nulle et d’une dilution infinie, qui concernent, quant à elles, plus particulièrement la viscosité intrinsèque. Les mesures rhéologiques peuvent être effectuées de diverses manières, notamment par la détermination de la contrainte subie par le milieu en réponse à une déformation constante ou, au contraire, par la sollicitation de façon constante du milieu et examen de la déformation engendrée.Trois grandeurs peuvent être obtenues au terme d’une expérience de rhéologie:– la viscosité ( 兀), qui est le rapport de la contrainte de cisaillement à la vitesse de cisaillement;– le module de conservation (G ), qui est le rapport de la composante en phase de la contrainte sur la déformation sinusoïdale;– le module de perte (G ), qui est le rapport de la composante déphasée de 900 de la contrainte sur cette même déformation sinusoïdale.G caractérise l’élasticité du matériau alors que G traduit la dissipation de l’énergie par friction.Dans leur très grande majorité, les polymères ne se comportent pas comme des liquides newtoniens, lesquels sont caractérisés par une viscosité constante indépendante de la vitesse de cisaillement appliquée. Un liquide polymère manifeste, en réalité, toutes les caractéristiques d’un liquide pseudoplastique, c’est-à-dire qu’après un premier plateau newtonien sa viscosité aura tendance à baisser à mesure que la vitesse de cisaillement augmente. Aux vitesses de cisaillement les plus faibles, les chaînes présentent des conformations peu différentes de celle de l’état non perturbé et il est probable que les enchevêtrements contribuent pleinement à la résistance à l’écoulement. Avec l’augmentation de la vitesse de cisaillement, les macromolécules s’alignent dans la direction du cisaillement, prennent une forme anisotrope et se contractent dans la direction perpendiculaire à l’axe du cisaillement. Une autre caractéristique des liquides polymères est leur propension à se comporter comme des liquides thixotropes: à vitesse de cisaillement constante, leur viscosité diminue en effet avec le temps d’application de la contrainte.À l’instar du temps d’application de la contrainte ou de la vitesse de cisaillement, la concentration du polymère dans le milieu peut aussi grandement affecter la viscosité.Au-delà d’une certaine concentration d’espèce de masse molaire (Mc), appelée masse molaire critique, la viscosité peut augmenter de façon abrupte, traduisant la présence d’interactions intermoléculaires (enchevêtrements). Même s’il n’existe aucune théorie capable de prévoir l’apparition du phénomène d’enchevêtrement, on peut en donner une explication empirique. Tant que la masse molaire d’un échantillon est inférieure à Mc, la viscosité est essentiellement gouvernée par le coefficient de friction (f ). La viscosité est dans ce cas proportionnelle à C[ 兀]. Au-delà de Mc et en présence d’enchevêtrements, la coopérativité que requièrent les longues chaînes pour se mouvoir l’une par rapport à l’autre freine leur diffusion; la viscosité varie alors en C 憐 聾w.L’existence d’enchevêtrements peut ainsi être décelée à travers la mesure de G’ qui, dans son augmentation avec la fréquence de la déformation sinuso 練dale, marque un plateau avant de croître à nouveau. La masse molaire (Me) à partir de laquelle ces enchevêtrements se manifestent est empiriquement 1/2 Mc. On dit que les enchevêtrements sont des points de réticulation temporaire.Interprétation théorique du comportement rhéologiqueLe premier modèle proposé est celui de Rouse (1953). Il représente la chaîne comme un assemblage de perles reliées entre elles par des ressorts. Ces perles sont censées interagir par friction (avec un coefficient f ) avec leur milieu environnant, tandis que les ressorts répondent de façon élastique à une sollicitation. Le milieu visqueux exerçant une force d’entraînement sur la chaîne (force proportionnelle à la vitesse des perles), la solution des équations de vitesse des perles donne accès aux temps de relaxation caractéristiques des modes internes de mouvement de la chaîne. En réalité ni ce modèle ni sa version améliorée, qui tient compte de la force exercée par les unités répétitives adjacentes ainsi que du mouvement brownien de la chaîne, ne prennent en compte les enchevêtrements. Ces modèles, qui ignorent également la variation de la viscosité avec la vitesse de cisaillement, sont en fait bien appropriés pour les milieux dilués.La description qui répond le mieux aux réalités auxquelles est confronté l’expérimentateur est celle proposée par de Gennes à travers son concept de reptation [cf. GENNES (P.-G. de)]. Selon ce modèle, la chaîne confrontée aux obstacles que constituent les enchevêtrements se meut à l’intérieur d’un tube qui représente approximativement son contour, comme le ferait un serpent à travers les obstacles sans les franchir vraiment. À côté de ce mouvement longitudinal de désengagement du tube, le modèle envisage également la possibilité de changements conformationnels à l’intérieur même du tube. Cette théorie prédit que la viscosité est très dépendante de la concentration du milieu et de la masse molaire de l’échantillon ( 兀 théorique est proportionnelle à M3 alors que 兀 mesurée est proportionnelle à M3,4). Même si résultats expérimentaux et prédictions théoriques ne concordent pas toujours très bien, la meilleure preuve de la réalité du phénomène de reptation est à trouver dans la différence entre les polymères linéaires et les polymères ramifiés, ces derniers diffusant effectivement beaucoup plus lentement.Quatre régions de viscoélasticité en fonction de la températureSi l’étude du comportement des solutions de fluides polymères est déterminante pour la connaissance des caractéristiques moléculaires d’un échantillon, pour sa mise en forme, pour sa rhéologie ou encore pour la réalisation de quelques applications bien ciblées, il est aussi essentiel de cerner les propriétés des polymères à l’état solide pour pouvoir les utiliser comme matériaux.À une température suffisamment basse, tout échantillon polymère est rigide, dur et se déforme de façon essentiellement élastique (dans la limite des faibles déformations), indépendamment du type d’organisation qu’adoptent ses chaînes: organisation totalement amorphe ou semi-cristalline. Le fait que certaines chaînes ne sont pas en mesure de cristalliser est essentiellement dû aux irrégularités et aux imperfections de leur structure. À mesure que la température s’accroît, le polymère a tendance à se comporter comme un matériau viscoélastique, réunissant les caractéristiques d’un liquide visqueux et celles d’un solide élastique. C’est à travers l’évolution des propriétés mécaniques que les phénomènes de viscoélasticité peuvent être le mieux mises en évidence. Dans un tel matériau viscoélastique, les propriétés mécaniques, et en particulier le module d’élasticité, dépendent de la durée d’application de la contrainte et de la température à laquelle s’applique la contrainte. Deux phénomènes caractéristiques se manifestent:– la relaxation, qui correspond à la diminution de la contrainte nécessaire pour maintenir une déformation constante au cours du temps;– le fluage, qui traduit l’augmentation de la déformation à contrainte constante.La relaxation se réfère, au plan moléculaire, au temps que mettent les macromolécules à répondre à une sollicitation extérieure par un mouvement d’orientation dans la direction de la contrainte. Quant au fluage, il résulte du glissement des chaînes les unes par rapport aux autres à tension constante.Quatre régions principales peuvent être distinguées au cours de l’étude viscoélastique d’un matériau polymère:– La première région correspond à l’état vitreux (état solide). Le module d’élasticité est de l’ordre de 3 憐 109 Pa indépendamment de la nature chimique du polymère. Les mouvements portent sur des distances très courtes et se limitent à la vibration, voire à la rotation d’éléments locaux de la chaîne. Des relaxations secondaires, concernant en particulier des groupes latéraux à la chaîne principale, ont pu être mises en évidence sur des polymères tels que le poly(méthacrylate de méthyle).– La deuxième région est celle de la transition vitreuse. Plusieurs théories ont été proposées pour rendre compte du phénomène de transition vitreuse. La première est celle dite du volume libre, selon laquelle le volume de matière inoccupée (ou libre) est invariable au-dessous de la température de transition vitreuse (Tg). Un deuxième groupe de théories décrit la transition vitreuse comme un phénomène purement cinétique qui se manifeste dès lors que le temps de réponse à une sollicitation est du même ordre de grandeur que le temps caractéristique de l’expérience. Enfin la théorie de Gibbs-Di Marzio fonde l’existence de la transition vitreuse sur le postulat que l’entropie de conformation est égale à O à Tg, même si les auteurs reconnaissent l’aspect cinétique du phénomène. Cependant, aucune de ces théories n’est vraiment en mesure d’expliquer de façon satisfaisante la transition vitreuse. Quoi qu’il en soit, la température à laquelle intervient cette transition dépend de la nature chimique de la chaîne macromoléculaire. Le module de conservation G’ décroît d’un facteur 103 pour une variation de 20 à 30 0C: la région de la transition vitreuse peut être qualitativement définie comme celle où apparaissent des mouvements coordonnés sur de longues distances.– La troisième région de viscoélasticité est celle du plateau caoutchoutique. Le polymère présente le comportement typique d’un caoutchouc avec des mouvements coordonnés sur de longues distances. Le module d’élasticité prend des valeurs de l’ordre de 2 憐 106 Pa et le plateau caoutchoutique est d’autant plus étendu que les chaînes sont longues. Si l’échantillon est réticulé, ce plateau caoutchoutique peut s’étendre sur une grande plage de température.– La quatrième et dernière région est celle de l’écoulement. À des températures plus élevées encore, l’énergie fournie permet aux chaînes de serpenter à travers les enchevêtrements et de s’écouler.Le comportement observé pour les échantillons semi-cristallins diffère quelque peu de celui des matériaux amorphes. Comme la température de fusion des parties cristallines est toujours supérieure à celle de la transition vitreuse dans les polymères semi-cristallins, le module d’élasticité prend des valeurs qui dépendent du degré de cristallinité des chaînes. Au-delà de la température de fusion des zones cristallines, le polymère s’écoule à l’instar d’un matériau amorphe.Cette partie concernant les propriétés viscoélastiques des matériaux polymères à l’état solide ne saurait être complète sans que soit évoquée le principe d’équivalence temps-température. Selon ce principe, le temps et la température sont deux grandeurs équivalentes pour les matériaux viscoélastiques. A partir de portions de courbe donnant l’évolution du module de conservation G’ en fonction du temps et à différentes températures, il est ainsi possible de tracer une courbe dite maîtresse à une température de référence par translation et superposition de ces différentes parties de courbe. Cette courbe maîtresse serait impossible à établir pour la majorité des polymères, par exemple à température ambiante, pour des raisons techniques: la plage de fréquences de sollicitation disponibles sur les appareillages est en effet restreinte. Le comportement d’un matériau viscoélastique sollicité à température élevée pendant des temps courts est ainsi comparable à celui du même matériau à basse température et pendant des temps plus longs. En d’autres termes, un changement de température produit le même effet que l’application d’un facteur multiplicatif à l’échelle des temps.Bien que jeune, la science des polymères a connu un développement constant depuis les travaux de H. Standinger dans les années 1920, qui s’est concrétisé par une multiplicité d’applications dans des domaines très divers. De nouvelles avancées ne pourraient résulter que d’une fructueuse collaboration entre chimistes et physiciens.5. Les macromolécules biologiquesTous les êtres vivants sont constitués de cellules, d’une seule, comme chez les bactéries ou les algues primitives; ou de plusieurs, environ 1015, comme chez les mammifères. Il est généralement admis que tous les êtres vivants dérivent d’une seule cellule primitive, apparue sur la Terre il y a environ 3,5 milliards d’années. Cette cellule s’est reproduite et les descendants ont évolué. Aujourd’hui, il existe deux grands types de cellules, la cellule eucaryote, qui possède un noyau, et la cellule procaryote, qui n’en possède pas. Ces cellules présentent beaucoup d’autres différences, comme la taille, l’organisation, la complexité, etc., mais se ressemblent par la nature des substances chimiques qu’elles contiennent et par leurs quantités relatives (tabl. 1). Le constituant principal des cellules est l’eau. En plus des ions inorganiques, les autres constituants, tous formés d’un squelette carboné [cf. ORGANIQUE (CHIMIE)], peuvent être divisés en deux grands groupes en fonction de leur masse molaire. Le premier groupe comprend des molécules de faible masse molaire inférieure à 2 000 grammes par mole (limite arbitraire). Ce sont essentiellement les sucres, les acides gras, les acides aminés, les nucléotides et tous les précurseurs et intermédiaires du métabolisme – au total, près de 750 espèces différentes. Le second groupe comprend les molécules de grande masse molaire appelées macromolécules ou biopolymères. Les biopolymères (de l’ordre de 2 000 espèces différentes dans une bactérie) sont les protéines, les acides nucléiques et les polysaccharides, formés par l’union de motifs élémentaires ou monomères que sont les acides aminés, les nucléotides et les sucres, respectivement. Les lipides et les phospholipides ne sont pas des macromolécules, bien qu’ils puissent s’associer et former de très grosses particules. Dans une macromolécule, les monomères (identiques ou différents) sont liés entre eux par des liaisons covalentes (interactions fortes), alors que l’association des phospholipides et des lipides résultent d’interactions beaucoup plus faibles (d’origines électrostatique et hydrophobe).Les biopolymères ont un rôle essentiel dans le fonctionnement des cellules. Les progrès considérables réalisés dans tous les domaines scientifiques et techniques ont permis d’isoler un grand nombre de macromolécules biologiques et de déterminer, de façon très précise, leurs masses molaires, leurs formes globales et locales (dans certains cas à l’échelle atomique) et leurs fonctions biologiques.Les acides nucléiquesDeux familles d’acides nucléiques, l’acide désoxyribonucléique (ADN) et l’acide ribonucléique (ARN), sont présentes dans les cellules. L’ADN et l’ARN résultent de l’enchaînement linéaire régulier de nucléotides (séquences). Chaque nucléotide est constitué d’une base hétérocyclique azotée (adénine, guanine, cytosine ou thymine dans le cas de l’ADN; adénine, guanine, cytosine et uracile dans le cas de l’ARN), d’un sucre et d’un groupement phosphate. du point de vue chimique, la différence principale, apparemment minime, entre les nucléotides de l’ADN et l’ARN porte sur la nature du sucre, un désoxyribose et un ribose, respectivement. Néanmoins, l’ADN et l’ARN ont des structures et des fonctions tout à fait différentes.Dans la cellule, l’ADN adopte une structure en double hélice droite, résultant de l’association non covalente de deux brins (ou chaînes) complémentaires; chaque nucléotide d’un brin étant apparié à un nucléotide de l’autre brin. Cette structure a été décrite pour la première fois, en 1953, par F. H. C. Crick et par J. D. Watson. La masse molaire de l’ADN, très grande, varie suivant les espèces. L’ADN humain contient 3 milliards de paires de nucléotides. Si cet ADN pouvait être complètement étiré, il aurait une longueur d’environ 2 mètres pour un diamètre de l’ordre de 2 憐 10—9 mètres. De nombreuses études sur des fragments d’ADN, naturels ou synthétiques, ont démontré le polymorphisme de l’ADN. Suivant les conditions expérimentales, un fragment d’ADN adopte une structure en double hélice droite (avec des variantes dans les paramètres hélicoïdaux) ou une structure en double hélice gauche ou même des structures hélicoïdales plus complexes contenant trois ou quatre brins. Aussi bien sur une paire de nucléotides que sur plusieurs centaines ou milliers de nucléotides, la structure de l’ADN est dynamique et non statique.L’ADN est le support de l’information génétique (dans certains cas comme les rétrovirus, l’information génétique est portée par l’ARN, mais l’insertion de cette information dans l’ADN de la cellule hôte ne se fera qu’après copie de l’ARN en ADN par une enzye, la transcriptase inverse). Les ordres qui déterminent la nature et le nombre de la quasi-totalité des macromolécules intervenant dans le métabolisme cellulaire proviennent de l’ADN et sont transmis par l’intermédiaire d’une autre macromolécule, l’ARN messager. L’information génétique est conservée intégralement au cours de la division de la cellule mère en deux cellules filles. La cellule est capable de synthétiser deux copies conformes de son ADN (si l’on excepte les mutations), grâce à la complémentarité des deux brins de la double hélice [cf. NUCLÉIQUES (ACIDES)]. Cependant, il serait erroné de penser que le rôle de l’ADN n’est qu’informatif. L’ADN matrice interfère, par sa structure dynamique, avec l’action de protéines comme celles qui sont impliquées dans la réplication (synthèse d’ADN) et dans la transcription (synthèse de l’ARN).La transcription de l’ADN en ARN donne naissance à trois grandes classes d’ARN qui englobent les ARN messagers, les ARN ribosomaux et les ARN de transfert (chez les eucaryotes, d’autres ARN interviennent dans le métabolisme nucléaire).Comme l’ADN, les ARN ont une structure bien définie mais, contrairement à l’ADN, cette structure ne met pas en jeu l’association de plusieurs chaînes. Chaque chaîne d’ARN se replie sur elle-même avec appariement de nucléotides complémentaires situés à plus ou moins grande distance le long de la chaîne (modèle multi-épingle à cheveux ou en multi-tige boucle). Le repliement tridimensionnel de la chaîne dépend de la séquence nucléotidique et conduit donc à une structure propre à chaque chaîne d’ARN.Les trois classes d’ARN ont des fonctions différentes mais leur couplage dirige la synthèse protéique. La synthèse d’une protéine nécessite l’assemblage entre un ARN messager (qui porte l’instruction définissant la séquence de la protéine qu’il code), les ribosomes (édifices formés par l’association des ARN ribosomaux et de protéines permettant l’enchaînement des acides aminés) et les ARN de transfert (chaque sorte d’acide aminé étant fixé à un ARN de transfert spécifique).Pendant longtemps, il a été accepté que seules les protéines possédaient des propriétés catalytiques. Dans les années 1980, on a montré que des séquences d’ARN sont capables de catalyser la coupure et la formation de liaisons covalentes entre nucléotides. Leur existence permet d’expliquer plusieurs processus biologiques, et ouvrent ainsi de nouvelles perspectives dans la compréhension des phénomènes prébiotiques, c’est-à-dire ceux qui ont permis l’origine de la vie [cf. ORIGINE DE LA VIE]: l’ARN serait apparu avant l’ADN et aurait joué à la fois les deux rôles de porteur de l’information génétique et d’enzyme. Ces ARN catalytiques, appelés ribozomes, sont aussi efficaces dans le tube à essai et en absence de protéines.Les protéinesLes protéines constituent une classe de macromolécules d’une grande complexité. Elles résultent de l’enchaînement linéaire régulier d’acides aminés. Vingt sortes d’acides aminés, différant par la nature chimique de leurs chaînes latérales, entrent dans la composition des protéines, d’où une très grande diversité des protéines (l’enchaînement de 100 acides aminés différents permet de générer 20100 protéines). Les masses molaires des protéines couvrent un très large domaine, mais en général sont inférieures à 100 000 grammes par mole. En se basant sur la forme globale, on peut diviser les protéines en deux grands groupes: les protéines globulaires (sphères ou ellipsoïdes) et les protéines fibreuses (bâtonnets). Dans tous les cas, la composition en acides aminés et la séquence de la protéine déterminent la structure et la fonction de la protéine. Comprendre comment une protéine adopte une structure tridimensionnelle fonctionnelle fait encore à l’heure actuelle l’objet de très nombreux travaux.Le repliement d’une protéine résulte d’interactions faibles (non covalentes) entre les acides aminés. Ces interactions faibles stabilisent localement des structures ordonnées (hélice 見 et feuillet 廓) qui contiennent un nombre variable d’acides aminés. Les hélices 見 et les feuillets 廓 interagissent entre eux pour former des unités globulaires (domaines) qui à leur tour peuvent interagir entre elles. Enfin, plusieurs protéines (deux ou plus, identiques ou pas) peuvent s’associer pour former des édifices complexes actifs de grandes masses molaires. Il est remarquable que la structure tridimensionnelle des protéines, stabilisée par des interactions faibles, soit aussi précise. Le remplacement d’un acide aminé par un autre (mutagenèse) peut entraîner la modification de la structure de la protéine. Souvent, les protéines subissent des modifications post-traductionnelles spécifiques, comme la phosphorylation ou la fixation covalente de groupes carbohydrates (ces protéines sont alors appelées des glycoprotéines) ou d’acides gras (lipoprotéines).Les protéines sont les agents actifs de la cellule. Elles interviennent à tous les niveaux (rigidité structurale, perméabilité des membranes, transport, concentration des métabolites, déplacement, fonctionnement des gènes, etc.). Certaines d’entre elles, appelées enzymes, ont la propriété remarquable d’être des catalyseurs spécifiques. Un catalyseur augmente la vitesse d’une réaction sans changer ni la direction de la réaction ni le rapport à l’équilibre entre les produits et les réactifs et se retrouve intact à la fin de la réaction. Les enzymes sont capables d’augmenter la vitesse d’une réaction de 106 à 1012 fois par rapport à celle de la même réaction non catalysée et dans les mêmes conditions expérimentales. De plus, elles sont très spécifiques, en ce sens qu’elles sont capables de différencier des substrats de formules chimiques voisines. Enfin, les réactions sont souvent coordonnées sous l’action de complexes multienzymatiques. Les enzymes permettent de réaliser, dans une bactérie, plus de 3 000 réactions chimiques différentes, intégrées dans un ordre spatial et temporel précis et controlé.PolysaccharidesLes polysaccharides résultent de l’enchaînement linéaire ou ramifié de sucres. Bien qu’une quinzaine de sortes de sucres entrent dans la composition des polysaccharides, chaque polysaccharide ne contient qu’un nombre limité (de 1 à 6) de sortes de sucres. Tout en ayant la même composition chimique, des polysaccharides peuvent être différents. C’est le cas de l’amylose, de l’amylopectine et du glycogène, qui sont des polymères de l’ 見-D-glucopyranose. L’un de ces polymères – qui diffèrent par la position des liaisons covalentes entre les monomères – est linéaire (amylose), les deux autres sont ramifiés (amylopectine et glycogène). Contrairement aux acides nucléiques et aux protéines, qui ont presque toujours une masse molaire définie, les masses molaires des polysaccharides sont souvent variables. Les polysaccharides peuvent se structurer mais pas de façon aussi remarquable et variée que les protéines. En plus de leur position, l’orientation des liaisons covalentes entre monomères dans des homopolymères conduit à des structures différentes. L’amylose a une structure lâche alors que la cellulose a une structure très ordonnée. Toutes ces possibilités dans l’enchaînement des monomères rendent très compliquée la caractérisation physico-chimique des polysaccharides.Certains polysaccharides servent de stocks de matières nutritives. C’est le cas du glycogène dans les cellules animales et de l’amidon dans les cellules végétales: ces réserves de glucides sont décomposables lors du métabolisme (glycolyse). D’autres polysaccharides interviennent dans la construction des parois des cellules végétales et bactériennes. Enfin, de courts polysaccharides (oligosaccharides) greffés sur certaines macromolécules jouent un rôle très important dans les phénomènes de signalisation qui entrent en jeu au cours de l’activité cellulaire.Remarques sur les macromolécules biologiquesDe nombreuses études sont encore consacrées à l’élucidation de la structure et de la fonction de biopolymères dans des milieux physiquement simples (solutions diluées, cristaux...). L’intérêt de ces études s’est trouvé conforté parce que, en parallèle, il est vérifié que des réactions se produisant dans la cellule peuvent être reproduites, au moins partiellement, dans le tube à essai en utilisant un mélange adéquat de biopolymères purifiés et de petites molécules. Il est ainsi possible de répliquer et de transcrire l’ADN, de synthétiser des protéines, toutes ces réactions obéissant aux lois classiques de la physique et de la chimie. Cependant, les équilibres réactionnels ainsi que les vitesses des réactions peuvent être complètement modifiés dans la cellule. En effet, le milieu cellulaire est hautement organisé; les biopolymères sont confinés dans un espace restreint (d’où une concentration élevée) et les réactions sont couplées. Dans des solutions très concentrées de complexes ADN-protéines (conditions se rapprochant de celles qui sont rencontrées dans le noyau des cellules eucaryotes, bien que les concentrations ioniques, le pH et le degré d’hydratation soient peu connus), d’une part les propriétés fonctionnelles des complexes sont stimulées et, d’autre part, les interactions entre ces complexes donnent naissance à une organisation cristalline liquide. Il est certain que les problèmes d’organisation à l’échelle supramoléculaire dans les êtres vivants interviennent à plusieurs niveaux (membranes, cellule elle-même, tissus cellulaires, etc.) et jouent un rôle fondamental dans le fonctionnement biochimique.La complexité des êtres vivants est telle que, pour l’aborder, les scientifiques de différentes disciplines doivent mettre leurs compétences en commun. Réciproquement, la meilleure connaissance des propriétés des biopolymères et de leurs fonctions dans la cellule est à la base de la conception et de la synthèse de systèmes de plus en plus complexes, présentant des propriétés ressemblant de mieux en mieux à celles du vivant.Les études physico-chimiques des macromolécules biologiques ont joué un rôle prépondérant dans l’impressionnant développement des sciences biologiques durant la seconde moitié du XXe siècle. Ce développement entraîne des bouleversements profonds pour l’homme et son environnement. Chaque découverte ouvre de nouvelles possibilités de la maîtrise du vivant. En 1996, pour la première fois dans l’histoire des sciences, un mammifère a été créé à partir du noyau d’une cellule somatique prélevée sur un animal adulte. Être capable de cloner un mammifère adulte a une portée considérable, tant du point de vue fondamental qu’appliqué, mais ouvre aussi la perspective de l’utilisation de ces techniques de reproduction à l’homme. Devant un problème d’une telle ampleur, une réflexion entre toutes les communautés est absolument nécessaire pour définir les droits et les devoirs de l’homme face à la science.
Encyclopédie Universelle. 2012.